Publié le 22 jan 2018Lecture 6 min
Le carcinome de Merkel
Astrid BLOM, CHU Ambroise-Paré, Boulogne-Billancourt
Le carcinome de Merkel est une tumeur agressive de plus en plus fréquente, dont le traitement au stade locorégional doit faire appel à la technique du ganglion sentinelle et à la radiothérapie adjuvante. Les avancées récentes sur la physiopathologie de cette tumeur viro-induite devraient permettre d’avoir prochainement des thérapies plus efficaces dans les stades avancés.
Le carcinome de Merkel est un carcinome neuroendocrine primitif cutané, rare et agressif. Il est plus fréquent chez les patients âgés, immunodéprimés et/ou s’étant beaucoup exposés au soleil, ce qui explique son incidence croissante actuelle. Sa classification en stades AJCC (American Joint Committee on Cancer) a été récemment mise à jour pour mieux appréhender son pronostic. Son étiopathogénie est mal élucidée, mais fait intervenir dans la majorité des cas le polyomavirus du carcinome de Merkel. L’immunité cellulaire est un élément-clé de la réponse antitumorale et des résultats prometteurs ont été obtenus dernièrement par l’immuno thérapie dans le traitement des stades avancés.
Incidence
Le carcinome de Merkel est rare. Il atteint presque exclusivement les patients caucasiens. Son incidence est en augmentation rapide depuis plusieurs années dans tous les pays développés. En France, l’incidence standardisée est actuellement estimée à 0,4 pour 100 000 personnes/an.
Clinique
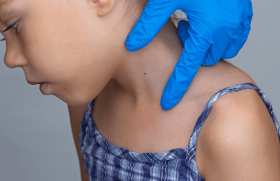
Le carcinome de Merkel est plus fréquent chez l’homme que chez la femme et se localise préférentiellement à l’extrémité céphalique ou aux membres supérieurs, plus rarement aux membres inférieurs ou au tronc. L’aspect clinique de la tumeur est très variable : il peut s’agir d’une tumeur violacée exophytique, parfois saignotante, ou à l’inverse d’une masse souscutanée avec une peau normale en regard (figures 1 et 2). Par analogie avec les critères ABCDE du mélanome, l’acronyme AEIOU a été proposé pour définir les caractéristiques cliniques principales du carcinome de Merkel :
- A pour asymptomatique (tumeur habituellement indolore) ;
- E pour évolution (tumeur augmentant rapidement en taille) ;
- I pour immunosuppression (environ 10 % des patients sont immunodéprimés, le risque de développer un carcinome de Merkel est multiplié par 10 en cas de greffe d’organe, par 13 en cas de séropositivité VIH et par 30 en cas de leucémie lymphoïde chronique) ;
- O pour « old age » (âge médian au diagnostic de 75 ans) ;
- et U pour UV (la majorité des lésions survient en peau photoexposée).
Dans environ 10 % des cas, aucune tumeur primitive cutanée n’est retrouvée, le diagnostic étant alors posé sur une métastase ganglionnaire.
Figure 1. Carcinome de Merkel se présentant sous la forme d'une tumeur violacée exophytique.
Figure 2. Carcinome de Merkel se présentant comme une masse sous-cutanée avec peau normale en regard.
Éthiopathogénie
Environ 80 % des carcinomes de Merkel en Europe et en Amérique du Nord sont associés au Merkel cell polyomavirus (MCPyV). Ce nouveau virus, décrit pour la première fois en 2008, est présent de façon normale sur la peau saine de la majorité des sujets adultes. Sous l’effet de stimuli mal élucidés, le génome viral intègre de façon clonale l’ADN d’une cellule de Merkel et exprime l’antigène sT et une forme tronquée de l’antigène LT. Ces deux oncoprotéines virales perturbent les mécanismes du cycle cellulaire (notamment en inactivant Rb), entraînant la prolifération tumorale. Dans la minorité de carcinomes de Merkel MCPyV-négatifs (15-20 %), la tumorogenèse est probablement médiée par des altérations de plusieurs voies de signalisation cellulaire (PI3K/AKT, TP53, etc.) par le biais de mutations UV-induites multiples.
Diagnostic
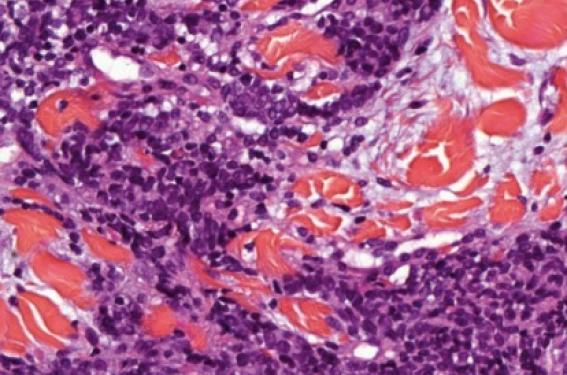
Le diagnostic est fait par l’exa men anatomopathologique. Il montre une prolifération dermique épargnant l’épiderme, faite de petites cellules au cytoplasme peu abondant et au noyau rond et hyperchromatique, pouvant envahir profondément le tissu sous-cutané pour atteindre le fascia et le muscle. Les mitoses sont fréquentes, de même que les images d’invasion angiolymphatique. L’immunohistochimie permet de poser le diagnostic, la tumeur étant positive à la fois pour les marqueurs neuroendocrines (synaptophysine, chromogranine A, neuron-specific enolase) et les marqueurs épithéliaux (AE1/AE3, pan-kératine). Une image pathognomonique est le marquage en point juxtanucléaire de la cytokératine 20, quasi-constant (figure 3). Enfin, le thyroid transcription factor 1 est habituellement négatif, permettant d’éliminer une métastase de carcinome bronchique à petites cellules.
Figure 3. L'aspect histologique du carcinome de Merkel : une prolifération de petites cellules rondes avec un marquage en point juxta-nucléaire par l'anti-CK20.
Classification et pronostic
Le stade au diagnostic est défini selon la classification TNM de l’AJCC. Celle-ci a été modifiée récemment (tableau ci-dessous) afin de mieux classer le groupe des patients découverts au stade de métastase ganglionnaire (stade III). En effet, la survie de ces patients varie selon la présence ou non d’un primitif cutané identifié au diagnostic, la survie à 5 ans étant multipliée par 2 en cas de primitif inconnu. Les principaux facteurs pronostiques sont la présence d’une immunosuppression (les patients immunodéprimés ont 2 fois plus de risque de mourir de leur carcinome de Merkel) et le stade de la maladie (survie à 5 ans de 63 % pour les stades I et seulement 13 % pour les stades IV).
Prise en charge initiale
Un examen clinique complet comprenant la palpation des aires ganglionnaires doit être pratiqué. Un bilan d’extension comprenant une échographie ganglionnaire et un scanner thoraco-abdomino-pelvien, voire un PET-scanner, est également recommandé.
Aucun examen sanguin n’a démontré un intérêt dans le bilan d’extension et le suivi du carcinome de Merkel. À l’avenir, il est possible que les dosages des cellules tumorales circulantes et/ou des anticorps contre les antigènes T de MCPyV puissent être des éléments de pronostic et de suivi (non validés actuellement et disponibles uniquement dans les centres spécialisés).
Tout patient présentant un carcinome de Merkel doit être discuté en réunion de concertation pluridisciplinaire. Il existe désormais en France un réseau national pour la prise en charge des carcinomes de Merkel, le réseau CARADERM, avec des centres experts référents dans chaque région (www.caraderm.org).
Traitement
Des recommandations de traitement françaises, européennes (voir http://sfdermato.org/recommandations-scores-etechelles/recommandations.html) et américaines existent pour le carcinome de Merkel et sont relativement superposables. En l’absence d’adéno pathie palpable, elles reposent dans un premier temps sur une exérèse élargie de la tumeur avec marges de 1 à 2 cm allant en profondeur jusqu’au fascia. Dans le même temps opératoire, une recherche du ganglion sentinelle doit être proposée, celui-ci étant envahi dans près d’un tiers des cas, même en cas de primitif de petite taille. En cas de ganglion sentinelle envahi ou d’adénopathie macroscopique, un curage ganglionnaire est recommandé.
Dans tous les cas, le traitement chirurgical doit être complété par une radiothérapie adjuvante du site du primitif (50 à 60 Grays). Celle-ci peut également être discutée sur l’aire de drainage ganglionnaire selon les cas : au décours du curage en cas de ganglion positif, ou en l’absence d’exérèse du ganglion sentinelle en cas de tumeur primitive à haut risque (grande taille, invasion angiolymphatique, patient immunodéprimé).
Perspectives thérapeutiques
Aucun traitement consensuel n’existe pour le carcinome de Merkel au stade métastatique. La chimiothérapie (sels de platine + VP16) est efficace (55 % de réponses), mais avec un échappement rapide (3 mois environ) et une toxicité non négligeable dans cette population âgée.
Étant donné le lien étroit dans cette pathologie entre immunité cellulaire et réponse antitumorale (évolution plus agressive chez les patients immunodéprimés, pronostic plus favorable des stades III en cas de régression spontanée de la tumeur primitive, meilleure survie en cas de présence de lymphocytes T CD8+ intratumoraux), il paraissait logique de proposer dans cette indication un traitement par immunothérapie. C’est désormais chose faite avec la publication récente des résultats de 2 essais cliniques princeps. Le premier a évalué un anti-PDL1, l’avelumab, en 2e ligne chez des patients ayant un carcinome de Merkel stade IV ayant échappé à la chimiothérapie. Parmi les 88 patients évalués, le taux de réponses objectives était de 32 % (9 % de réponses complètes) et la survie sans progression à 6 mois de 40 %. La 2e étude a évalué un anti-PD1, le pembrolizumab, en 1re ligne dans 26 cas de carcinomes de Merkel métastatiques ou inopérables. Le taux de réponses objectives était de 56 % (16 % de réponses complètes) et la survie sans progression de 67 % à 6 mois. Ces résultats très encourageants pourraient permettre dans un avenir proche l’autorisation de mise sur le marché de ces molécules dans le carcinome de Merkel.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :