Maladie de système, Médecine interne
Publié le 11 avr 2007Lecture 16 min
Quels syndromes se cachent derrière une tumeur annexielle ?
J. KANITAKIS Clinique dermatologique, Hôpital Édouard-Herriot, Lyon
Les tumeurs annexielles de la peau sont des tumeurs bénignes, beaucoup plus rarement malignes, qui dérivent de (ou se différencient vers des) structures annexielles cutanées, follicules pileux, glandes sébacées et glandes sudorales (eccrines ou apocrines). Elles sont souvent uniques et isolées ; lorsqu’elles sont multiples, elles peuvent avoir un caractère familial et être la manifestation cutanée de syndromes génétiques comportant des néoplasies viscérales. Il est donc important pour le dermatologue de connaître ces tumeurs, qui sont souvent révélatrices de syndromes potentiellement graves sur le plan pronostique.
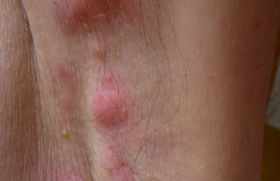
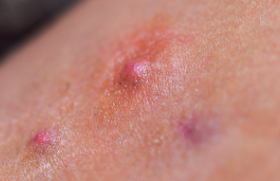
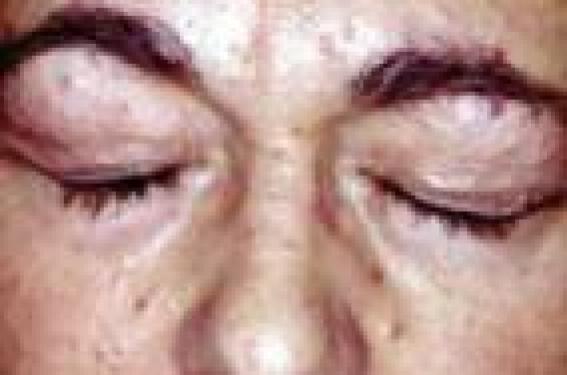
Le syndrome de Cowden Trichilemmomes et autres signes cliniques • Les trichilemmomes (TC) sont des tumeurs bénignes dérivant de la gaine folliculaire externe de la partie profonde (infra-infundibulaire) des follicules pileux, appelées également trichilemmes. Il s’agit cliniquement de papules à surface lisse ou verruqueuse du visage, souvent périorificielles (nez, bouche), de 1-4 mm (figure 1). Figure 1. Syndrome de Cowden : multiples papules faciales (trichilemmomes). • Histologiquement, les TC forment un nodule bien limité, en léger relief sur le plan cutané, constitué d’une prolifération de kératinocytes volumineux à cytoplasme clair PAS-positif (figure 2). Lorsqu’ils sont multiples, les TC sont un marqueur du syndrome de Cowden (SC). Figure 2. Aspect histologique d’un trichilemmome : nodule dermique bien limité, constitué d’une prolifération de kératinocytes à cytoplasme clair (hématoxyline-éosine). Lorsqu’ils sont multiples, les trichilemmomes sont un marqueur du syndrome de Cowden. • Ce syndrome, du nom de la première famille décrite par Lloyd et Denis en 1963, est une génodermatose autosomique dominante à pénétrance et expression variables. • Outre les TC, lésions cutanées les plus fréquentes (83 % des patients), le SC comporte une papillomatose gingivale, labiale et linguale (donnant à la muqueuse gingivale un aspect « pavimenteux »), des papules kératosiques du dos des mains et des pieds (43 %), des papules kératosiques translucides palmo-plantaires (42 %) sans caractère histologique spécifique. Ces lésions apparaissent au cours de la 3e-4e décennie de la vie et précèdent habituellement l’apparition des lésions viscérales. Plus rarement, on peut observer des lipomes, des hémangiomes, une langue scrotale, des tumeurs nerveuses (schwannomes, neuromes), des fibromes, des xanthomes, un vitiligo, des taches café au lait, des tumeurs à cellules de Merkel, des mélanomes et des carcinomes cutanés. • Les manifestations extracutanées du SC, qui en font sa gravité, concernent le sein (maladie fibrokystique ou carcinomes, atteignant 76 % des femmes), l’intestin (40 % des cas : hamartomes, polypose), la thyroïde (67 % ; goître, adéno(carcino)mes), le tractus urogénital (55 % ; kystes ovariens, léiomyomes, cancers endométriaux), le squelette (37 % ; macrocéphalie, cyphose, voûte palatine) et le système nerveux (19 % ; retard mental, épilepsie, maladie de Lhermitte-Duclos, anomalies de l’EEG, médulloblastomes). Globalement, des cancers sont observés chez 52 % des femmes et 23 % des hommes atteints de SC. Diagnostic Des critères diagnostiques ont été mis au point par l’International Cowden Consortium en 2000 et sont fondés sur la présence de différents signes, classés en pathognomoniques (trichilemmomes, kératoses acrales, papillomatose muqueuse), majeurs (cancers du sein, de la thyroïde et de l’endomètre, macrocéphalie, maladie de Lhermite-Duclos) et mineurs (autres lésions thyroïdiennes, retard mental, hamartomes gastro-intestinaux, maladie fibrokystique des seins, lipomes, fibromes, tumeurs génitales). Diagnostic du syndrome de Cowden Le diagnostic de SC est posé, si les conditions suivantes sont réunies : • 6 (ou plus) papules faciales, dont 3 au moins correspondent à des TC histologiquement documentés ; • papillomatose muqueuse associée à des papules faciales ou à des kératoses acrales ; • au moins 6 kératoses palmo-plantaires ; • maladie de Lhermitte-Duclos ou macrocéphalie associée à un critère majeur ; • association d’un critère majeur et 3 critères mineurs ; • présence de 4 critères mineurs ; • antécédents familiaux de SC associés à un critère pathognomonique ou majeur ou à 2 critères mineurs. Devant une atteinte cutanée évocatrice de SC, il faudra rechercher des tumeurs extracutanées par un examen clinique (dermatologique et gynécologique) complet, une échographie thyroïdienne, une coloscopie, des mammographies, un frottis vaginal, une enquête familiale à la recherche de cas similaires, voire une IRM cérébrale en présence de signes cliniques évocateurs d’une atteinte cérébrale. Aspects génétiques Le SC est transmis selon le mode autosomique dominant, avec une pénétrance et expression variables. Il est dû à des mutations du gène suppresseur de tumeurs PTEN (Phosphatase and TENsin homologue), localisé sur le chromosome 10q23-3. L’importance des mutations du gène PTEN dans le développement du SC est démontrée par l’existence d’un modèle animal : les souris « knock-out » homozygotes pour ce gène (PTEN -/-) décèdent in utero, alors que des animaux « knock-out » hétérozygotes (PTEN +/-) développent des néoplasies multiples, réalisant un tableau équivalent à celui du SC chez l’homme. Des mutations du gène (PTEN) sont également à l’origine du syndrome de Protée et du syndrome de Bannayan-Riley-Ruvalcaba, et il a été proposé de regrouper ces entités sous le terme de « syndrome des hamartomes multiples/ tumeurs associé à des mutations du PTEN ». Traitement Le traitement des lésions cutanées du SC repose sur l’utilisation de rétinoïdes, de 5-FU, la cryothérapie et l’exérèse chirurgicale ou au laser à CO2. Un traitement général par rétinoïdes peut être essayé, bien que l’effet préventif sur l’apparition de tumeurs cutanées soit empirique en l’absence d’études. La surveillance de patients atteints de SC n’est pas codifiée. Il est licite de surveiller les organes ayant un fort risque de dégénérescence par un examen gynécologique, des mammographies, des frottis cervicaux annuels, une échographie thyroïdienne annuelle, des examens endoscopiques digestifs. Le syndrome de Muir-Torre Adénomes sébacés et autres signes cliniques • Les adénomes sébacés sont des tumeurs papulo-nodulaires jaunâtres ou brunâtres, parfois cratériformes ou d’aspect kystique, siégeant préférentiellement sur le visage (figure 3). Histologiquement, ils sont constitués d’une prolifération de sébocytes plus ou moins matures formant des nodules dermiques de taille variable, parfois ulcérés (figure 4). Figure 3. Syndrome de Muir-Torre : multiples tumeurs jaunâtres ou brunâtres, parfois cratériformes du visage (adénomes sébacés). Figure 4. Aspect histologique d’un adénome sébacé : nodule dermique constitué d’une prolifération de sébocytes à différents stades de maturation (hématoxyline-éosine). Ils sont donc différents des hyperplasies sébacées séniles, lésions banales du visage chez des sujets d’un certain âge, parfois appelés à tort « adénomes sébacés ». La présence de tumeurs adénomateuses sébacées multiples doit faire suspecter le syndrome de Muir-Torre (SMT) caractérisé par l’association de tumeurs cutanées et de tumeurs viscérales uniques ou multiples. Le syndrome de Muir-Torre est caractérisé par l’association de tumeurs cutanées et de tumeurs viscérales. • Les lésions cutanées du SMT comprennent : – des adénomes sébacés (kystiques ou non) : il s’agit cliniquement de lésions papulo-nodulaires blanchâtres ou jaunâtres, uniques ou multiples, de 1-5 cm, siégeant sur l’extrémité céphalique dans 70 % des cas ; – des épithéliomas sébacés, qui simulent cliniquement les carcinomes basocellulaires ; – des véritables carcinomes sébacés, reconnus histologiquement par la présence d’atypies nucléocytoplasmiques et de mitoses ; – de kératoacanthomes qui peuvent présenter une différenciation sébacée, sont souvent multiples et apparaissent chez des sujets jeunes ou sur peau non exposée au soleil. • Les atteintes viscérales du SMT comprennent des cancers, qui sont multiples dans 40 % des cas : – cancers colorectaux (50-60 %), intéressant surtout la partie droite du côlon ; ils apparaissent à un âge relativement jeune et sont d’assez bon pronostic ; – cancers uro-génitaux (25 %), intéressant l’endomètre, les ovaires, la prostate, les reins, les uretères, la vessie ; – cancers du sein, des poumons, du larynx, de la parotide, de la langue, du système hématopoïétique (hémopathies). Des polypes intestinaux bénins ainsi que diverses tumeurs bénignes osseuses ou utérines peuvent également se voir. • Le SMT atteint un peu plus fréquemment les hommes que les femmes (sexe ratio : 3/2) et l’âge moyen au moment du diagnostic est de 55 ans. Dans 40 % des cas, les tumeurs cutanées sont diagnostiquées avant ou pendant le diagnostic de cancer viscéral. Dans 40 % des cas, les tumeurs cutanées sont diagnostiquées avant ou pendant le diagnostic de cancer viscéral. Aspects génétiques Sur le plan génétique, le SMT est transmis selon le mode autosomique dominant avec une expression et une pénétrance variables. Dans la majorité des cas, il est dû à des mutations germinales des gènes de réparation des erreurs d’appariement de l’ADN (MSH2 : 90 % ; MLH1 : 10 %), induisant une instabilité des microsatellites. Du fait des mutations, les protéines codées par ces gènes ne sont pas exprimées dans les tumeurs sébacées, ce qui peut être détecté par immunohistochimie sur coupes histologiques, et contribuer au diagnostic (figure 5). Figure 5. Syndrome de Muir-Torre : absence d’expression de la protéine MSH2 dans un adénome sébacé (haut de la figure), qui est normalement exprimée dans un follicule pilosébacé normal (bas de l’image) (immunohistochimie, révélation aminoéthylcarbazole). L’anomalie génétique est identique à celle du syndrome de Lynch (HNPCC, cancer colorectal héréditaire non associée à une polypose), et le SMT est considéré comme une variante allélique du syndrome de Lynch. Un second type de SMT a été plus récemment identifié. Il n’est pas associé à des mutations de ces gènes et sa pathogénie demeure inconnue. Diagnostic Le diagnostic du SMT repose sur l’association de différents critères, qui sont classés en trois groupes (groupe A : cancer viscéral ; groupe B : adénome, épithélioma, carcinome ou kératoacanthome sébacé ; groupe C : kératoacanthomes multiples, cancers viscéraux multiples ou antécédents familiaux de SMT). Le diagnostic est posé en présence du critère A et d’au moins un critère du groupe B ou de trois critères du groupe C (en l’absence de facteurs cancérigènes connus, par exemple : radiothérapie précédente, maladie de Kaposi, lymphomes, SIDA). • Le diagnostic différentiel doit se faire avec les syndromes de Cowden, de Gardner, de Gorlin, les kératoacanthomes multiples spontanément régressifs (Fergusson-Smith), la sclérose tubéreuse de Bourneville. Explorations carcinologiques Si le diagnostic de SMT est suspecté, un bilan doit être réalisé à la recherche orientée de cancer par coloscopie, frottis aspiratif endométrial, échographie abdo-pelvienne et mammographie, biopsies des tumeurs cutanées et viscérales (côlon), dosage de marqueurs tumoraux sériques (ACE), recherche d’hémorragies digestives occultes, radiographie thoracique. Les patients doivent faire l’objet d’une surveillance comportant coloscopie tous les 2 ans après l’âge de 25 ans, ou à réaliser 5 ans avant l’âge du cas familial le plus précoce connu. Une consultation gynécologique annuelle doit être pratiquée après l’âge de 30 ans avec mammographie et frottis aspiratif. Un bilan urinaire (cystoscopie, échographie) et digestif (fibroscopie, échographie) est conseillé tous les 2 ans. Traitement Les traitements locaux des lésions du SMT reposent sur l’exérèse chirurgicale (qui doit être large en cas de carcinome sébacé), la cryothérapie, le laser CO2, voire le 5-FU intralésionnel pour les kératoacanthomes. L’isotrétinoïne (0,5 mg/kg/j) peut être utilisée pour stabiliser les lésions sébacées, mais des rechutes se produisent à l’arrêt du traitement. Les cancers viscéraux seront traités de façon appropriée (chirurgie, chimiothérapie). Le syndrome de Gardner/polypose adénomateuse familiale • Les kystes folliculaires (épidermoïdes ou trichilemmaux) sont des tumeurs banales observées le plus souvent sur le cuir chevelu. Lorsqu’ils sont multiples, familiaux et surtout lorsqu’ils comportent des signes histologiques inhabituels (kystes hybrides, associant des aspects de kyste épidermoïde et trichilemmal, voire pilomatriciel) (figure 6) ils peuvent constituer un marqueur du syndrome de Gardner (SG), syndrome transmis selon le mode autosomique dominant ; le gène responsable est nommé APC (Adenomatous polyposis coli), siège sur le locus 5q21, et ses mutations sont également la cause de la polypose adénomateuse familiale, dont le SG est considéré comme une variante phénotypique. Figure 6. Kyste hybride, comportant une paroi en partie épidermoïde (à gauche) et une partie pilomatricielle (à droite) (hématoxyline-éosine). • Les manifestations cutanées du SG comportent des kystes épidermoïdes ou trichilemmaux multiples, et surtout des kystes « hybrides », présents sur le visage, le cuir chevelu et les membres dans 50-60 % des cas, des tumeurs desmoïdes post-traumatiques de la paroi abdominale, plus rarement des fibromes, des lipomes, des léiomyomes ou des neurofibromes. • Les manifestations extracutanées comportent des polypes rectocoliques multiples (50-65 %), dont la transformation maligne est quasi constante vers l’âge de 40 ans, des ostéomes de la mandibule ou du crâne (> 50 %), des lésions pigmentées multifocales rétiniennes (80 %), congénitales ou se développant très tôt dans la vie, des anomalies dentaires (18 % : anodontie, odontomes, dents surnuméraires ou rudimentaires, caries multiples), et parfois d’autres carcinomes (thyroïde, ovaires, hépatoblastome, craniopharyngiome, médulloblastome, glioblastome, ostéo- ou liposarcome). Les lésions oculaires sont congénitales ou se développent très tôt dans la vie, et les lésions cutanéo-osseuses précèdent la polypose. • Le diagnostic différentiel du SG doit se faire avec les kystes épidermoïdes multiples simples, la polypose adénomateuse familiale (qui ne comporte ni kystes ni lésions osseuses) et le syndrome de Turcot associant polypose colique, taches café au lait et tumeurs malignes du système nerveux central. • En cas de suspicion d’un SG, le bilan doit comporter une coloscopie, des radiographies du tube digestif et du squelette, un examen du fond d’œil, la recherche d’hémorragie digestive occulte, une enquête familiale. • Le traitement repose sur l’exérèse chirurgicale des kystes et des ostéomes, et une exérèse des polypes par endoscopie, voire une colectomie préventive. Le syndrome de Birt-Hogg-Dubé • Les trichodiscomes et les fibrofolliculomes sont des tumeurs histologiquement apparentées, caractérisées par une prolifération du mésenchyme entourant les follicules pileux. Cliniquement, il s’agit de petites papules blanchâtres siégeant sur le visage, le cou et plus rarement le tronc. L’association de (multiples) fibrofolliculomes, trichodiscomes et acrochordons (molluscum pendula) caractérise le syndrome de Birt-Hogg-Dubé (SBHD), syndrome génétique rare, de transmission autosomique dominante. • Le SBHD est dû à des mutations du gène BHD1, gène suppresseur de tumeurs localisé sur le chromosome 17p11.2 qui code pour une protéine nommée folliculine, présente dans la peau et les poumons. • Le SBHD apparaît vers l’âge de 25 ans. Le diagnostic est fait en présence d’au moins 10 fibrofolliculomes (ou trichodiscomes) associés à des acrochordons. Les manifestations extracutanées du SBHD comprennent : – des tumeurs rénales (risque relatif > 9), coliques, thyroïdiennes, parotidiennes (oncocytome), des (angio)lipomes, des tumeurs nerveuses et conjonctivales ; – des pneumothorax spontanés à répétition, un emphysème, des kystes pulmonaires (risque relatif : 32). La surveillance doit se faire par des radiographies thoraciques et une échographie rénale annuelle à vie. •Le diagnostic différentiel du SBHD doit se faire avec les syndromes de Cowden, de Muir-Torre et de Gardner. La sébocystomatose • Les kystes sébacés sont une variété particulière de kystes d’origine folliculaire, comportant une paroi épithéliale épidermoïde dans laquelle sont incluses des glandes sébacés. Ils ne doivent pas être confondus avec les kystes trichilemmaux, beaucoup plus fréquents, parfois abusivement dénommés « kystes sébacés ». La présence de nombreux kystes sébacés définit la sébocystomatose (ou steatocystoma multiplex), génodermatose autosomique dominante apparaissant à l’adolescence, caractérisée par la présence de dizaines voire de centaines de papulo-nodules asymptomatiques jaunâtres ou rougeâtres, à contenu huileux ou laiteux, plus ou moins malodorant, fréquemment compliqués de poussées inflammatoires. Ces lésions siègent sur le thorax, le dos, le visage, le cuir chevelu, la racine des bras et des cuisses. • Le traitement est symptomatique et repose sur l’incision ou l’ablation des kystes au bistouri ou au laser (YAG ou CO2). L’isotrétinoïne par voie orale peuvent stabiliser les lésions et diminuer la fréquence des poussées inflammatoires, mais leur arrêt est suivi de récidives. La cylindromatose et le syndrome de Brook-Spiegler • Les cylindromes sont des tumeurs nodulaires siègeant avec prédilection sur le cuir chevelu. Ils sont caractérisés histologiquement par une prolifération de massifs tumoraux fortement basophiles, à contours nets, agencés entre eux comme des pièces d’un « puzzle » ; chaque massif est entouré d’une membrane basale hyaline épaisse PAS-positive (figure 7). Figure 7. Aspect histologique d’un cylindrome : proliferation de massifs épithéliaux basophiles entourés d’une épaisse membrane basale (hématoxyline-éosine). Ces tumeurs sont volontiers multiples, réalisant un aspect de tumeur « en turban » ; elles sont souvent familiales et peuvent être associés à d’autres tumeurs annexielles comme les trichoépithéliomes, les grains de milium et les spiradénomes eccrines siégeant sur le visage et le tronc, réalisant alors le syndrome de Brooke-Spiegler (SBS) ou syndrome de multiples cylindromes/trichoépithéliomes. • Ce syndrome est transmis selon le mode autosomique dominant et est associé à des mutations du gène CYLD-1 localisé sur le chromosome 16q21-23. Les lésions apparaissent pendant l’enfance ou la 2e décennie et augmentent progressivement en nombre et en taille. • Le traitement repose sur l’excision chirurgicale au bistouri ou au laser CO2. Autres syndromes Il faut enfin signaler qu’il existe d’autres syndromes génétiques complexes qui comportent des tumeurs annexielles ; cependant dans ces cas, les tumeurs cutanées sont rarement à l’origine du diagnostic car les symptômes extracutanés sont prépondérants : grains de milium mutiples du visage et des mains dans le syndrome oro-digito-facial de type 1, pilomatricomes multiples dans le syndrome de Rubinstein-Taybi et la myotonie dystrophique de Steinert, hamartome sébacé dans le syndrome de Schimmelpenning, trichoépithéliomes multiples dans le syndrome Rombo, syringomes multiples dans la trisomie 21, hidrocystomes des paupières dans le syndrome de Schopf-Schutz-Passagre. Conclusion Plusieurs tumeurs annexielles sont la manifestation cutanée de syndromes génétiques complexes, dont certains comportent un risque de cancers viscéraux. Ces tumeurs sont essentiellement les trichilemmomes (syndrome de Cowden), les adénomes sébacés (syndrome de Muir-Torre), les fibrofolliculomes/trichodiscomes (syndrome de Birt-Hogg-Dubé), les kystes épidermoïdes ou hybrides (syndrome de Gardner). L’apparition des tumeurs annexielles précède souvent celle des tumeurs viscérales observées au cours de ces syndromes. Le dermatologue doit donc connaître ces associations afin de pouvoir diagnostiquer précocement ces « syndromes à cancer ».
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :