Psoriasis
Publié le 11 mai 2022Lecture 9 min
Florent AMATORE, Emmanuel DELAPORTE, Service de dermatologie, Hôpital Nord, Marseille

Le psoriasis pustuleux (PP) diffère du psoriasis commun en plaques à la fois sur le plan clinique, épidémiologique et physiopathologique, si bien que de nombreux auteurs s’accordent à dire qu’il s’agit en réalité de deux maladies différentes. Le PP regroupe lui-même plusieurs entités distinctes mais qui ont en commun une présentation clinique caractéristique faite de pustules superficielles non folliculaires et amicrobiennes, liées à une accumulation intraépidermique de polynucléaires neutrophiles. La récente identification d’anomalies génétiques et de mécanismes immunologiques sous-jacents a permis des progrès dans sa compréhension et dans sa prise en charge thérapeutique.
Généralités
Le psoriasis pustuleux (PP) inclutplusieurs sous-types, d’évolution et de sévérité variables, qui peuvent survenir isolément ou être associés à un psoriasis classique. La classification du PP est controversée. En 2017, le European Rare and Severe Psoriasis Expert Network (ERASPEN) a émis un consensus sur l’individualisation de trois phénotypes cliniques de PP : une forme généralisée (psoriasis pustuleux généralisé [PPG]) et deux formes localisées (psoriasis pustuleux palmoplantaire, acrodermatite continue de Hallopeau [ACH])(1). En revanche, les ouvrages historiques francophones et anglophones de dermatologie distinguent, au sein des formes généralisées, le PPG érythrodermique (dit de von Zumbusch), le PPG exanthématique (forme peu sévère et transitoire), le PPG lié à la grossesse (improprement appelé impétigo herpétiforme), le PPG infantile et le PPG annulaire(2).Plusieurs phénotypes peuvent survenir chez un même individu, mais cela n’est pas classique. Du fait de la rareté et de l’hétérogénéité du PP, il n’existe pas de données épidémiologiques robustes. Il semble toutefois plus fréquent dans les populations asiatiques que caucasiennes, et les formes généralisées semblent toucher des patients plus jeunes (3e décennie) que les formes palmoplantaires et l’ACH (5e décennie)(3).
Phénotypes cliniques
PPG érythrodermique (von Zumbusch)
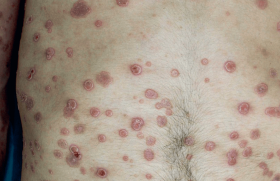
Il s’agit de la forme la plus sévère. Elle est caractérisée par l’apparition brutale d’un érythème généralisé, parfois œdémateux, qui se couvre rapidement de multiples pustules, de couleur blanc laiteux, qui peuvent confluer en vastes placards annulaires (figure 1). L’état général est altéré, avec une fièvre importante, des frissons, des nausées et des arthralgies. Une atteinte des muqueuses (pustules sublinguales, érosions, chéilite, conjonctivite) peut être associée. Le tableau évolue spontanément vers une desquamation diffuse en lambeaux scarlatiniformes et une régression des signes généraux, et de nouvelles poussées identiques surviennent après quelques jours ou semaines, pouvant mettre en jeu le pronostic vital du patient.
Sur le plan biologique, on observe classiquement une hyperleucocytose à PNN, un syndrome inflammatoire, une cytolyse hépatique, une hypocalcémie et une hypoalbuminémie. À l’histologie, on trouve une pustule stérile située à la partie supérieure du corps de Malpighi.
On y voit les PNN s’immiscer entre les kératinocytes qui perdent leur noyau et prennent un aspect de réseau dont les mailles contiennent les PNN. Cela explique le terme de « pustulose spongiforme multiloculaire » (dite de Kogoj-Lapière). Les similitudes cliniques et histologiques sont nombreuses avec la pustulose exanthématique aiguë généralisée (PEAG). La prise récente d’un médicament imputable ou un épisode infectieux concomitant, l’absence d’antécédent de psoriasis ou de survenue de psoriasis au décours de la poussée, la présence d’une vasculite histologique sont des arguments en faveur de la PEAG, mais tous ces critères sont discutables, et seule une meilleure compréhension des anomalies immunologiques qui sous-tendent ces deux affections permettra de préciser leur degré de différence(4).La mortalité du PPG érythrodermique est élevée, allant jusqu’à 30 % selon les séries(5). Les principales causes de décès sont le sepsis, les défaillances respiratoires et cardiaques, en plus des complications classiques d’une érythrodermie. Les nouvelles thérapeutiques (voir dans « traitement ») offrent toutefois un espoir considérable.
PPG lié à la grossesse (impétigo herpétiforme)
Cette entité, rare, a été décrite pour la première fois en 1872 par Ferdinand von Hebra. Le tableau clinico-biologique est identique à celui du PPG érythrodermique, avec un début plus souvent dans les plis et une généralisation secondaire. Les signes généraux sont majeurs, avec parfois des signes neurologiques (convulsion, myalgies,parésies...) liés à l’hypocalcémie. Il survient lors du 3e trimestre de grossesse ou juste après l’accouchement, et récidive lors de grossesses ultérieures, avec souvent une sévérité plus importante et un début plus précoce(6). Des cas de récidives lors de la prise d’une contraception œstroprogestative ont également été décrits. Le pronostic maternel (défaillance multiviscérale) et fœtal (retard de croissance, mort fœtale) est réservé.
PPG annulaire
Il consiste en une éruption de larges placards érythémateux annulaires ou polycycliques, d’évolution centrifuge, dont la périphérie est couverte de micropustules qui laissent rapidement place à un liseré desquamatif (figure 2). Les signes systémiques sont légers ou absents. Il touche des sujets plus jeunes, et son évolution, spontanément favorable, et de bien meilleur pronostic que le PPG érythrodermique.
PPG infantile
Il est extrêmement rare et n’a été décrit que dans des petites cohortes. Des cas familiaux ont été rapportés. Il survient dans la première année de vie ou aux alentours de 6 ans et peut se présenter soit sous une forme généralisée, avec une clinique semblable à celle du PPG érythrodermique de l’adulte, soit sous une forme annulaire. Les récidives sont fréquentes, et le pronostic est incertain.
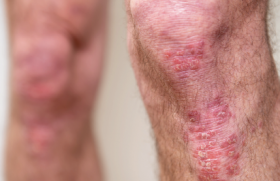
PP palmo-plantaire
Il touche les paumes et/ou les plantes de manière bilatérale et symétrique (figure 3). Il se présente sous la forme de pustules jaunâtres coalescentes sur un fond érythémateux et hyperkératosique. Parfois, les pustules se kératinisent formant un bloc adhérant en « clou de tapissier ». Les éminences thénar, hypothénar et la partie interne de la voûte plantaire sont les sites les plus souvent atteints, mais des formes diffuses à l’ensemble de la paume ou de la plante sont possibles. Le PP palmo-plantaire affecte plus souvent les femmes d’âge moyen et les fumeurs(7). L’association avec une atteinte articulaire est fréquente, jusqu’à 30 % dans les séries japonaises(8). Il peut également s’intégrer à des maladies auto-inflammatoires telles que le syndrome SAPHO.
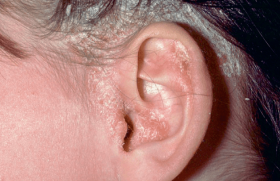
Acrodermatite continue de Hallopeau
L’ACH est rare et touche avec prédilection les femmes d’âge moyen. Elle se manifeste par l’évolution chronique de multiples pustules de l’extrémité distale des doigts ou des orteils, qui reposent sur une plaque rouge brillant et desquamative. L’atteinte du pouce est classique, et tous les doigts peuvent être touchés simultanément. Les poussées successives entraînent une atrophie dermo-épidermique et une destruction de l’ongle (figure 4). L’os de la phalange sous-jacente peut être atteint, pouvant aller jusqu’à l’ostéolyse. Comme pour le PP palmo-plantaire, l’association à une atteinte articulaire est fréquente.
Physiopathologie
Malgré un certain degré de similitude et de chevauchement entre PP et psoriasis classique, il existe des différences dans les mécanismes physiopathologiques qui sous-tendent les deux affections. Le PP résulte d’une cascade inflammatoire impliquant une hyperactivité des cellules de l’immunité innée, avec un rôle central de la voie de l’IL-36, alors que le psoriasis classique fait plutôt appel à l’immunité adaptative et à l’axe IL-17/IL-23(3).
L’IL-36 est une cytokine pro-inflammatoire qui fait partie de la superfamille de l’IL-1, une cytokine incontournable dans la réponse inflammatoire innée. Elle comprend 3 sous-unités agonistes (IL-36α,-β,-γ) et une antagoniste (IL-36RA).
L’IL-36 est exprimée par de nombreux types de cellules immunitaires mais aussi par les kératinocytes. Sécrétée sous forme de précurseur, elle nécessite l’action des PNN pour devenir biologiquement active. Lorsqu’elle se fixe à son récepteur (IL-36R et IL-1RAcP), lui aussi présent à la surface des cellules immunitaires et épithéliales dans la peau (mais aussi les poumons et le tube digestif), elle déclenche une cascade inflammatoire via les voies intracellulaires NK-kB et MAP-kinases(9). Il en résulte une production d’autres cytokines pro-inflammatoires et de chimiokines, une régulation positive des gènes de l’inflammation et une hyperprolifération kératinocytaire. L’IL-36 stimule également la voie de l’IL-17, et exerce une action synergique avec l’IL-17A et le TNF-α(9).
Comme pour le psoriasis classique, la prédisposition génétique est un socle obligatoire pour le déclenchement d’un PP et de nombreux variants alléliques ont été identifiés chez les patients atteints de PPG. Les principaux gènes impliqués dans le PPG sont l’IL-36RN (qui code pour l’IL-36Ra, un antagoniste de l’IL-36), CARD14, AP1S3, MPO, SERPINA3 et TNIP1(3). Ainsi, des pertes de fonction du gène IL-36RN, responsables d’un défaut d’inhibition physiologique de l’IL-36, sont retrouvées chez plus d’un quart des patients atteints de PPG, et sont donc associées à un début plus précoce et plus sévère de la maladie(10). Les patients atteints d’une mutation monogénique nulle de l’IL-36RN ont un déficit de l’IL-36Ra (syndrome DITRA) et présentent un phénotype particulièrement sévère de PPG qui débute dans l’enfance ou à l’adolescence. Dans les formes localisées de PP, les anomalies génétiques sous-jacentes sont moins bien identifiées. Les variants pathologiques de l’IL-36RN ne concernent que 5 % des patients(11).
Comme pour le PPG, des mutations de AP1S3 et CARD14 ont été décrites mais ne semblent concerner qu’une faible proportion de patients. Dans le PP palmo-plantaire, l’inflammation de la glande sudoripare eccrine semble être le primum movens. Le rôle important des peptides antimicrobiens, notamment hCAP-18/LL37 dans l’induction de cette inflammation a récemment été souligné(12).
La prédisposition génétique n’est rien sans les facteurs environnementaux qui entretiennent la maladie. Stress, infections, grossesse, médicaments, sevrage rapide en corticoïdes systémiques sont les plus classiques. Dans l’actualité, des PPG ont été décrits après une infection par la Covid-19, et après une vaccination anti-Covid(13).
Des cas de PP paradoxal, surtout palmo-plantaires, sont fréquemment rapportés sous anti-TNFα. Enfin, le tabac semble jouer un rôle important dans le PP palmo-plantaire et l’ACH, probablement par l’augmentation des taux d’IL-17 et l’activation de voies inflammatoires médiées par la nicotine(14).
Traitement
La prise en charge du PP n’est pas facile, les échecs thérapeutiques sont fréquents. Les recommandations françaises publiées en 2019, qui sont en cours d’actualisation, hiérarchisent les différents traitements disponibles(15).
Dans le PPG, la ciclosporine et l’acitrétine sont préférés au méthotrexate. Parmi les biothérapies, l’infliximab et les anti-IL-17 ont un intérêt par rapport aux autres du fait de leur rapidité d’action. Plus récemment, le risankizumab et le guselkumab, deux inhibiteurs de l’IL-23, ont montré une franche efficacité dans des cohortes japonaises et sont en cours d’essai de phase III.
Mais c’est sans nul doute le spesolimab, un anticorps monoclonal anti-IL-36 récepteur, qui représente le principal espoir thérapeutique(16). Dans un essai randomisé de phase II versus placebo, incluant 53 patients dont 35 traités par spesolimab, les scores de « pustulation » et le Generalized pustular psoriasisphysician global assessment, qui vont de 0 (pas de pustules, peau blanchie) à 4, étaient évalués. Les patients recevaient une dose unique intraveineuse de 900 mg de spesolimab ou de placebo, suivie d’une ou de plusieurs doses en ouvert après le 8e jour en cas d’échec ou de récidive. Une semaine après la 1re dose, 54 % des patients du groupe spesolimab avaient un score de pustulation à 0, et 43 % un score GPPPGA à 0-1, contre respectivement 6 % et 11 % dans le groupe placebo. Deux patients traités par spesolimab ont présenté une allergie médicamenteuse et un peu moins de la moitié, un épisode infectieux. L’efficacité du spesolimab est donc prometteuse, avec une action spectaculairement rapide, mais de plus larges études sont nécessaires pour préciser son bénéfice sur le long terme et son risque vis-à-vis de l’immunodépression induite. Dans les formes localisées de PP, la ciclosporine et la PUVA locale, plus ou moins associée à l’acitrétine, offrent les meilleurs taux de réponse. Parmi les biothérapies, l’étanercept, l’ustékinumab et le guselkumab sont celles qui ont été le mieux évaluées.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :