Publié le 19 avr 2017Lecture 6 min
Acrodermatitis enteropathica : le point
Philippe BERBIS, Service de dermatologie, Hôpital Nord, CHU de Marseille
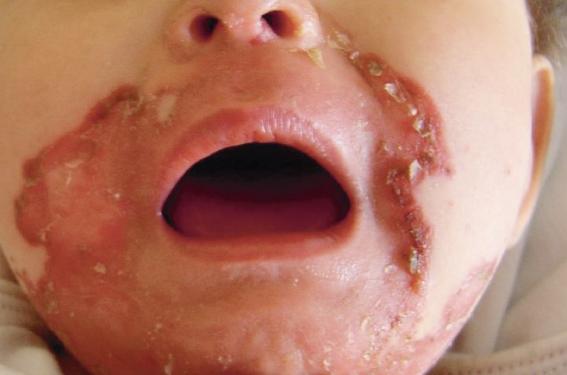
Une enfant de 13 mois était adressée par son pédiatre pour une dermatose érosive évolutive, touchant le cuir chevelu, le visage, le siège et les extrémités des membres, évoluant depuis 3 mois…
Observation
L’enfant était née à terme, nourrie au sein et sevrée depuis 4 mois. Elle était irritable, photophobique et présentait des troubles digestifs à type de diarrhée dont l’exploration s’était révélée négative.
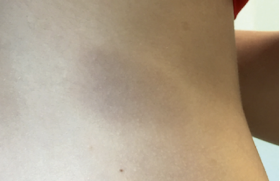
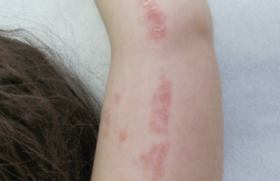
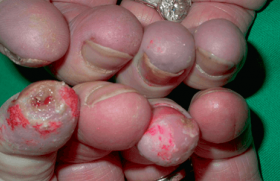
Des traitements topiques variés (antimycosiques, antibiotiques, dermocorticoïdes) n’avaient entraîné aucune amélioration, voire une aggravation. L’examen notait des plaques érosives étendues au niveau de la nuque, gagnant la région cervicale (figure 1), aux bords irréguliers, déchiquetés. Une alopécie diffuse, non cicatricielle était notée (figure 1). Des lésions érythémato-squameuses, bulleuses puis érosives étaient particulièrement nettes et évolutives au niveau péribuccal (figure 2). L’aspect devenait vite croûteux par endroits. L’atteinte périnéale était majeure (figure 3). Les extrémités des membres étaient touchées, bien qu’à un moindre degré, avec ici encore des lésions érythémato-squameuses, vaguement psoriasiformes et érosives par endroit (figure 4).
Figure 1. Lésions érosives diffuses du cuir chevelu et alopécie diffuse.
Figure 2. Lésions erythémato-squameuses et érosives péribuccales.
Figure 3. Érosions du pli interfessier.
Figure 4. Lésions psoriasiformes et érosives de la main.
Le diagnostic d’acrodermatitis enteropathica (AE) était évoqué, confirmé par la zincémie très abaissée (15 µg/dl pour une normale de 70 à 120 µg/dl). Le reste du bilan (NFS plaquettes notamment) était sans anomalie. Un traitement par sulfate de zinc entraînait en quelques jours une cicatrisation complète des lésions, la disparition des troubles digestifs et l’amélioration de l’état général et neurologique. La pousse des cheveux reprenait dès la fin du premier mois de traitement.
Discussion
L’AE a été identifiée en 1942, le lien avec la carence en zinc identifié en 1973 par Barnes et Moynahan. En 2002, la mutation du gène SLC39A4 était identifiée comme responsable de la maladie(1). Le zinc est un cation divalent, composant essentiel de plus de 300 métallo-enzymes et plus de 2 000 facteurs de transcription géniques. C’est donc un oligoélément important dans la synthèse protéique et celle de l’ADN. À ce titre, c’est un élément essentiel à la vie. Le zinc joue un rôle important dans les fonctions reproductives, les fonctions immunologiques, la cicatrisation, le fonctionnement du système nerveux. Cet oligoélément possède de plus des propriétés antioxydantes. Le zinc est principalement stocké dans le tissu osseux et musculaire, la peau contenant environ 6 % du zinc stocké dans l’organisme.
Le zinc est principalement absorbé au niveau du duodénum et du jéjunum, grâce à des transporteurs protéiques(2,3).
Il existe 2 types de transporteurs du zinc. Les transporteurs Znt (ou SLC30) réduisent les concentrations intracellulaires en zinc, alors que les transporteurs Zip (ou SLC 39) favorisent l’absorption du zinc par l’entérocyte. Il existe 15 protéines Zip.
Zip4 est le transporteur spécifique, assurant l’absorption active du zinc au niveau jéjunal. Le zinc peut aussi être absorbé par diffusion passive ou à l’aide de protéines riches en cystéine via l’attachement à la membrane apicale de l’entérocyte. Une fois absorbé par l’entérocyte, le zinc passe dans la circulation sanguine.
L’AE est une affection rare, sévère si non diagnostiquée précocement, transmise sur le mode autosomique récessif(4). Il n’y a pas de prédominance de sexe. Son incidence est évaluée à 1/500 000. La carence en zinc, quelqu’en soit la cause, si elle n’est pas compensée, entraîne une défaillance pluri-organique (tableau) pouvant conduire à de graves complications, notamment infectieuses, parfois mortelles, le zinc étant un oligoélément important dans le fonctionnement du système immunitaire, tant hu- moral que cellulaire. Le rôle du dermatologue est ainsi au premier plan compte tenu du caractère évocateur des manifestations cutanées. Le retard diagnostique, et donc thérapeutique, est particulièrement délétère chez ces enfants. Le gène muté est le gène CLC39A4(1), codant pour la protéine Zip4, situé sur le chromosome 8q24.3. Ce gène est composé de 12 exons, codant pour une protéine de 647 acides aminés.
Le début des symptômes apparaît lorsque l’enfant est sevré du lait maternel (qui contient des agents qui optimisent l’absorption du zinc), plus tôt chez les autres enfants, vers la 4e à la 10e semaine, lorsque les réserves en zinc sont éliminées. Les signes cutanés sont les plus évocateurs. Les lésions débutent par un érythème, une fragilité épidermique, puis des aspects bulleux, enfin érosifs, parfois impétiginisés. La topographie est très évocatrice : les lésions touchent prioritairement les zones périorificielles et les extrémités. L’atteinte du périnée est également constante, sous forme de placards érosifs douloureux. Les autres signes sont moins constants : alopécie diffuse non cicatricielle, onycholyse, stomatite, chéilite, blépharite, conjonctivite. Les signes microscopiques montrent une pâleur cytoplasmique des kératinocytes, une dégénérescence vacuolaire pouvant aller dans les formes avancées jusqu’à une nécrolyse épidermique superficielle. Une hypogranulose et une parakératose sont notées, expliquant l’aspect parfois psoriasiforme des lésions avancées. Le diagnostic d’AE nécessite un dosage du zinc plasmatique à jeun, qui doit être inférieur à 70 microgrammes/dl. L’étude de la mutation du gène SLC39A4 confirme le diagnostic. Ce gène a été cloné en 2002 par une équipe nantaise(1) qui a montré la responsabilité d’une mutation de ce gène dans la pathogénie de l’AE. SLC39A4 est régulée en fonction des apports alimentaires de zinc. Au plan clinique, le diagnostic différentiel peut se poser avec une épidermolyse bulleuse congénitale, une candidose sévère, une dermatite séborrhéique, une dermatite atopique, une histiocytose langerhansienne (notamment en cas d’atteinte sévère du cuir chevelu comme dans notre observation).
Le diagnostic différentiel des causes d’hypozincémie chez le nourrisson se pose peu : inadéquation de la substitution en zinc en cas d’alimentation parentérale, carence en zinc dans le lait maternel par mutation du gène SLC30A2 qui code pour la protéine ZnT2, transporteur impliqué dans la sécrétion du zinc dans le lait maternel(2). Dans ce dernier cas, la mise en évidence de taux faibles de zinc dans le lait maternel fait le diagnostic. La prématurité prédispose à la carence en zinc, pour des raisons multifactorielles (augmentation des besoins, immaturité digestive, réserves insuffisantes). Pour mémoire, chez l’adulte notamment, des carences en zinc avec des tableaux proches de l’AE peuvent s’observer dans des syndromes de malabsorption digestive, notamment au cours d’entéropathies inflammatoires chroniques, telle que la maladie de Crohn, de l’insuffisance pancréatique, de pathologies tubulaires rénales, et lors d’alimentations parentérales prolongées. Des carences vitaminiques (PP, B3) ou un syndrome du glucagonome peuvent donner des tableaux proches cliniquement de l’AE. Le traitement repose sur l’administration per os de zinc, sous forme de sulfate, à la posologie de 3 mg/kg/jour de zinc élémentaire. Le traitement doit être prolongé toute la vie. L’amélioration des signes cutanés est très rapide dès le début du traitement, en quelques jours. Un dosage de la zincémie doit être effectué tous les 3 à 6 mois. Le dosage de la cuprémie doit être effectué parallèlement. En effet, des zincémies trop élevées peuvent favoriser des hypocuprémies. Au total, le développement rapide de lésions érythémato-squameuses, vésiculo-bulleuses et érosives péribuccales et périnéales, associées à une alopécie et à une diarrhée, doit faire évoquer chez un nourrisson le diagnostic d’AE. Un diagnostic et un traitement rapide éviteront des complications parfois sévères.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :