Cancérologie
Publié le 08 déc 2009Lecture 6 min
Papulose lymphomatoïde : à propos de trois observations
Z. ALIOUA*, H. LAMSYAH*, N. HJIRA*, M. RIMANI**, N. BABA*, M. GHFIR*, O. SEDRATI* *Service de dermatologie **Service d’anatomie pathologique, Hôpital militaire d’instruction Mohammed V, Rabat, Maroc
La papulose lymphomatoïde (PLy) est une dermatose rare, d’étiologie inconnue, d’évolution chronique faisant actuellement partie des lymphomes T primitivement cutanés à évolution indolente (selon la classification EORTC [European Organisation for Research and Treatment of Cancer])(1). Elle se caractérise par le contraste entre un tableau clinique bénin, une image histologique d’apparence maligne et un potentiel de régression spontanée. Nous décrivons ici les caractéristiques anatomocliniques, thérapeutiques et évolutives de la papulose lymphomatoïde chez trois patients hospitalisés au service de dermatologie de l’hôpital militaire d’instruction Mohammed V de Rabat.
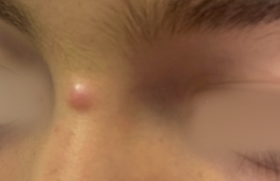
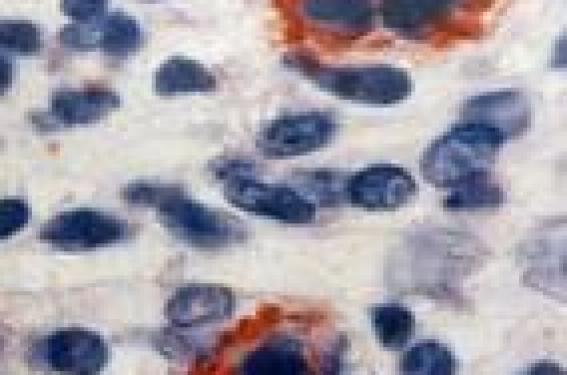
Notre expérience Observation n°1 Un patient de 41 ans, sans antécédents notables, est hospitalisé pour des lésions du tronc et des membres évoluant par poussées. À l’admission, l’examen cutané trouve des papulonodules de 0,5 à 1 cm de diamètre, recouverts de squames psoriasiformes ou à centre nécrotique. Le reste de l’examen somatique est sans particularité. L’examen anatomopathologique confirme le diagnostic de papulose lymphomatoïde type A avec présence de lymphocytes atypiques CD30 positif (figures 1 et 2). L’abstention thérapeutique a été recommandée. L’évolution a été favorable, marquée par la disparition des lésions avec un recul de 6 ans. Figure 1. Infiltrat dermique fait de lymphocytes. Figure 2. Lymphocytes atypiques CD30 positif. Observation n°2 Il s’agit d’un patient âgé de 42 ans, avec des antécédents de lèpre lépromateuse suivi dans notre service depuis 1990 et traité selon le protocole national, actuellement en observation sans traitement depuis 1995. En 2001, il a présenté des lésions papulonodulaires érythémateuses recouvertes par endroit de squames psoriasiformes, de 0,5 à 1 cm de diamètre, siégeant sur le tronc et les membres, avec respect du visage, des paumes, des plantes et des muqueuses. Le reste de l’examen somatique était sans particularité. Deux diagnostics ont été évoqués : une lèpre histoïde ou une papulose lymphomatoïde. L’histologie avec étude immunohistochimique a montré un infiltrat composé en majorité de lymphocytes T avec des cellules atypiques qui étaient de phénotype T et exprimant le CD30. Ceci a confirmé le diagnostic de papulose lymphomatoïde type A. Le patient a été mis sous méthotrexate (MTX) 20 mg IM par semaine (NFS et biologie hépatique normales). L’évolution a été favorable, marquée par l’affaissement des lésions dès la 4e injection et disparition totale des lésions au bout de 10 semaines, d’où arrêt du traitement après une dose cumulative de 200 mg de MTX. Le patient est toujours en rémission complète avec un recul de 4 ans. Observation n°3 Un patient de 45 ans, sans antécédents notables, présentait depuis 4 ans des lésions papulonodulaires érythémateuses dont quelques-unes étaient recouvertes de squames psoriasiformes, siégeant sur le tronc et les membres, le tout évoluant dans un contexte de conservation de l’état général. Les lésions évoluaient spontanément par poussées de 3 à 4 semaines, entrecoupées de rémissions d’environ 1 mois. L’examen anatomopathologique a confirmé le diagnostic de papulose lymphomatoïde type A avec présence de lymphocytes atypiques CD30 positif. Le patient a été mis sous MTX 20 mg IM par semaine, la rémission a été obtenue dès la 5e injection. Le malade a arrêté de lui-même le traitement. La récidive est survenue 1 mois après et le patient a été remis sous MTX à la même dose avec amélioration dès la 2e injection. Le traitement a été poursuivi avec une dose cumulative de 1 020 mg. L’évolution a été marquée de nouveau par la récidive à l’arrêt du MTX. Une photothérapie UVB (8 séances) a été instaurée. L’évolution a été favorable avec un recul de 4 ans. Discussion Décrite la première fois par W.L. Macaulay(2), la papulose lymphomatoïde appartient au spectre des proliférations lymphocytaires cutanées CD30+ dont elle représente 17,4 % des cas (3). Elle survient à tout âge (9-64 ans), mais surtout chez l’adulte jeune entre 20 et 50 ans, avec une prédominance masculine (sex-ratio H/F : 4/1) (3). Elle est caractérisée par des lésions maculopapulaires, parfois micronodulaires de 0,5 à 2 cm, de nombre variable et disséminées sur le corps. L’évolution se fait vers une croûte nécrotique mimant le pityriasis lichénoïde et disparaissant spontanément (3). Les lésions peuvent être prurigineuses. L’examen histologique montre un infiltrat dermique polymorphe constitué de petits lymphocytes, d’histiocytes, de polynucléaires neutrophiles et éosinophiles associés à quelques cellules de grande taille exprimant l’antigène CD30. Trois types de papuloses lymphomatoïdes ont été décrits : • la PLy de type A est caractérisée par une prolifération qui a la forme d’un triangle à base épidermique souvent nécrosée avec infiltrat dermique de grandes cellules exprimant le CD30/Ki-1 mêlées à des histiocytes, des polynucléaires neutrophiles et/ou éosinophiles et des lymphocytes normaux (1,4) ; • la PLy de type B, qui ne fait pas partie du spectre des proliférations lymphocytaires CD30+, se caractérise par un infiltrat épidermotrope constitué d’éléments lymphoïdes atypiques de type Sézary aux noyaux cérébriformes de taille petite à moyenne simulant un mycosis fongoïde et n’exprimant pas le CD30 (1,4-6). Les deux types A et B peuvent coexister chez le même patient ; • la PLy de type C se caractérise par un infiltrat monomorphe fait de nombreuses cellules CD30+ avec peu de cellules inflammatoires, ressemblant à un lymphome T à grandes cellules CD30+. Mais c’est essentiellement l’aspect clinique qui permet de distinguer une PLy d’un lymphome à grandes cellules CD30+ (7,8). L’évolution se fait par poussées de 2 à 4 semaines sur plusieurs années, avec des rémissions complètes pouvant être de longue durée. Le pronostic est généralement bénin avec un taux de survie à 5 ans de 100 %(3). La papulose lymphomatoïde a toujours suscité des discussions quant aux explorations nécessaires et aux traitements qu’il faut instaurer. Actuellement, on considère qu’il est inutile de réaliser un bilan d’extension. Si les lésions sont discrètes et peu symptomatiques, l’abstention thérapeutique est indiquée et les dermatocorticoïdes peuvent être ponctuellement utilisés (1,4,9). En cas de lésions plus diffuses et gênantes avec risque cicatriciel, une photothérapie (PUVAthérapie, UVB) peut être proposée. Une autre alternative est la chlorméthine (caryolysine) ou la carmustine (BCNU) en badigeonnage. Le méthotrexate à faible dose a aussi été proposé avec un effet durable (10). Nous avons opté pour ce traitement chez deux de nos patients devant la diffusion des lésions. Enfin, l’interféron a été utilisé très ponctuellement dans cette indication avec un bon résultat, mais sans données sur le long terme. Une surveillance prolongée de ces malades est nécessaire, car dans 10 à 20 % des cas, la PLy peut être associée ou suivie par un mycosis fongoïde, un lymphome CD30+ ou une maladie de Hodgkin (8,9). Conclusion La papulose lymphomatoïde, originale par sa clinique bénigne, son histologie inquiétante et son potentiel de régression spontanée, nécessite une attitude peu agressive adaptée à son bon pronostic. Toutefois, le suivi prolongé des malades est nécessaire du fait de la possibilité de transformation ou d’association à un lymphome cutané ou systémique, d’autant plus qu’il n’existe pas actuellement de critères pronostiques fiables permettant d’en prévoir la survenue.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :