Publié le 29 fév 2024Lecture 4 min
Hamartome épidermique : quelle prise en charge ?
Catherine FABER, d’après la présentation du Pr Smail Hadj-Rabia (Paris)
Un expert du Centre de référence des maladies rares de la peau et des muqueuses d’origine génétique (MAGEC) de l’hôpital Necker répond à trois questions sur les hamartomes épidermiques : quand explorer, quand biopsier et quel risque pour la descendance ?
L'hamartome est une malformation due à une anomalie du développement avant la naissance avec, au microscope, la présence dans l’organe d’une petite zone contenant des tissus qui ont, par eux-mêmes, un aspect normal mais sont disposés et associés de manière anarchique, ce qui leur enlève tout rôle fonctionnel. C’est une dysembryoplasie. En France, l’hamartome cutané désigne une tumeur bénigne non mélanocytaire et le terme « nævus » est réservé aux tumeurs cutanées bénignes mélanocytaires. Les hamartomes épidermiques (HE) ont bénéficié des progrès considérables réalisés ces dernières années dans les techniques d’analyse. On dispose aujourd’hui de techniques permettant de détecter dans un échantillon cutané une différence cellulaire, au niveau moléculaire, présente dans moins de 10 % des cellules(1). Ce type d’analyse peut être réalisé dans tous les tissus, à grande échelle et pour tout le génome en théorie.
Dans la démarche diagnostique de l’HE, il est important d’identifier la structure cutanée atteinte et de préciser l’arrangement des lésions ainsi que leur topographie et leur étendue. Les lésions sont souvent disposées selon les lignes de Blaschko qui correspondent aux lignes de migration des cellules cutanées embryonnaires. En fonction de la structure atteinte, on distingue les hamartomes kératinocytaires et les hamartomes annexiels ou organoïdes. La biopsie cutanée peut être utile en cas de doute avec un diagnostic différentiel. Elle est rarement utilisée pour faire un diagnostic précis d'hamartome.
NOUVELLES DONNÉES
L’amélioration des connaissances sur les syndromes d’HE a conduit à une mise à jour de leur classification en 2010(2) avec, entre autres, la définition d’une nouvelle entité, le syndrome CLOVES*. Les quelques exemples suivants illustrent les nouvelles informations disponibles. Dans l’hamartome comédonien, dû à une mutation du gène NEK9, il a été établi que, en cas de lésions très localisées, il n’y a pas lieu de s’inquiéter. Elles relèvent d’une exérèse totale. Des malformations associées, dont la plus fréquente est la cataracte, devront être recherchées essentiellement dans les formes étendues. Cette situation est extrêmement rare. Autre hamartome annexiel très rare, la phacomatose pigmento- kératosique – fraîchement rebaptisée phacomatose spilosébacée – se caractérise par un HE à différenciation sébacée, parfois kératinocytaire, associé à une tache café au lait et à un nævus mélanocytaire. Deux éléments apparaissent importants dans cet HE lié à une mutation du gène HRAS : ce sont aussi les formes étendues qui seront plus volontiers explorées et les anomalies associées sont souvent du même côté que l’HE. L’hamartome sébacé du syndrome de Schimmelpenning est le mieux connu et celui pour lequel des mutations de gènes de la voie des MAP-kinases (HRAS et KRAS) ont été décrites pour la première fois. La décision d’une exploration à la recherche de malformations associées est, là encore, guidée par la taille des lésions, mais aussi par la présence d’une atteinte céphalique.
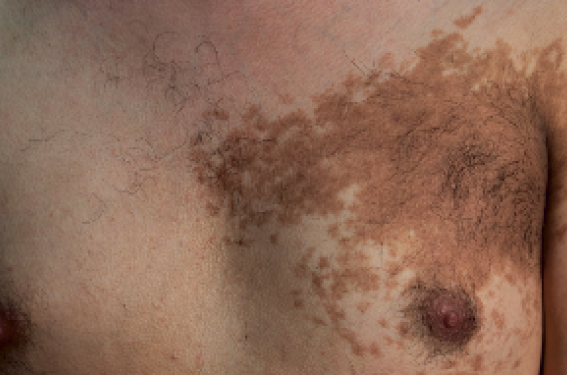
Enfin, les critères cliniques pré- valent aussi dans l’hamartome de Becker (photo). Il peut être syndromique et se manifeste cliniquement par des lésions unilatérales qui ne suivent pas les lignes de Blaschko, mais se présentent sous la forme d’une plaque hyperpigmentée et pileuse.
Des mutations de nombreux gènes, essentiellement de la voie des MAP-kinases, ont été associées aux HE kératinocytaires(3). Une petite proportion de patients (5 %) a non pas une, mais deux mutations. Les principaux signes extra-cutanés des syndromes d’HE sont neurologiques, ophtalmologiques et musculosquelettiques(2). Les traitements topiques ou par voie orale, comme dans le syndrome CLOVES*, la chirurgie et les lasers ablatifs(4) constituent les options thérapeutiques actuelles.
TRANSFORMATION MALIGNE ET RISQUE POUR LA DESCENDANCE
Concernant le risque d’évolution néoplasique évoqué pour certains HE, des données récentes font état d’une transformation maligne de 0,9 % des hamartomes sébacés du cuir chevelu, apparue le plus souvent à l’âge adulte(5). Les cancers internes sont surtout associés à des mutations HRAS. Il convient d’être attentif à la surveillance de la vessie chez les patients atteints d’hamartome pigmento-kératosique étendu. Par ailleurs, pour quasiment tous les syndromes d’HE, il n’y a pas de risque de transmission à la descendance. Celle-ci est possible quand la mutation est présente également dans les cellules gonadiques. Deux observations d’enfants nés avec une ichtyose épidermolytique, dont un parent avait un hamartome kératinocytaire épidermolytique, soulèvent la question(5,6,7). Une mutation de KRT10 a été détectée dans 3,9 % du sperme du père de l’un d’entre eux, et le second a hérité d’un nouveau variant du gène KRT1 de sa mère. Cette dernière avait un HE étendu.
En conclusion, les données sur les HE évoluent rapidement du fait des avancées importantes des techniques d’analyse. Une exploration est justifiée dans les formes étendues et en cas de lésions localisées sur le visage. De nouvelles possibilités thérapeutiques émergent.
* Congenital lipomatous over-growth, vascular malformations, epidermal nævi, scoliosis/skeletal and spinal syndrome.
D’après la présentation du Pr Smail Hadj-Rabia (Paris), Séminaire de dermatologie pédiatrique de l’hôpital Necker (SDPHN), 23 juin 2023.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :