Publié le 07 jan 2022Lecture 14 min
Les tumeurs cutanées malignes de l’enfant
Sylvie FRAITAG, Anatomo-cytopathologiste, hôpital Necker-Enfants malades, Paris

Les tumeurs malignes de l’enfant sont rares mais représentent une problématique majeure pour le clinicien. Le diagnostic précoce permettant la mise en route d’un traitement adapté est l’un des facteurs essentiels du pronostic. Ainsi, la réalisation d’une biopsie s’impose au moindre doute, en l’absence d’un diagnostic clinique certain.
Les tumeurs cutanées malignes sont rares chez l’enfant et ne représentent que 1,4 % des tumeurs ; les plus fréquentes étant représentées par les hamartomes et les tumeurs bénignes. Environ deux tiers des tumeurs malignes sont primitives, et un tiers sont métastatiques avec, par ordre de fréquence, le rhabdomyosarcome, les leucémies et le neuroblastome(1).
La répartition des tumeurs varie selon les tranches d’âge : les tumeurs cutanées malignes (TCM) sont plus fréquentes chez le nouveau-né et le nourrisson que chez l’enfant. Dans la période néonatale, on rencontre en majorité des leucémies (LA), des histicytoses langerhansiennes, des métastases de neuroblastomes, des sarcomes primitifs ou secondaires(2,3) . Ce sont toujours des urgences diagnostiques, les lésions cutanées pouvant être révélatrices d’une tumeur maligne viscérale ou d’une leucémie. Chez l’enfant prépubère, les tumeurs malignes les moins rares sont les sarcomes et les lymphomes ; le mélanome est exceptionnel. Les tumeurs malignes épithéliales ne s’observent que dans des syndromes de prédisposition à des cancers.
En période néonatale, la sémiologie clinique des lésions peut parfois permettre d’orienter vers une étiologie plutôt qu’une autre. Par exemple, une éruption papulo-nodulaire d’apparition explosive de couleur bleutée ou violacée ou un aspect de blueberry muffin baby doivent faire évoquer une leucémie aiguë, un neuro-blastome métastatique, voire une histiocytose langerhansienne(3) (tableau 1) ; une tumeur à consistance dure et à surface mamelon-née, un sarcome. La notion d’une génodermatose prédisposant aux tumeurs chez un « grand enfant » (xeroderma pigmentosum, nævo-matose basocellulaire) est une notion essentielle pour éliminer une TCM. Il en est de même en cas d’antécédent de radiothérapie.
Le diagnostic doit toujours être confirmé par l’examen histologique d’une biopsie cutanée, d’autant plus que certaines tumeurs bénignes peuvent être inquiétantes par leur taille (hamartomes conjonctifs, xanthogranulome juvénile néonatal) ou leur aspect (myofibromatose infantile), et peuvent mimer des tumeurs malignes. Inversement, certaines tumeurs malignes peuvent mimer des lésions bénignes (dermatofibrosarcomes congénitaux, tumeur fibrohistiocytaire plexiforme). Face à une tumeur de diagnostic non évident, il convient de faire une biopsie en prévoyant, en plus du fragment fixé, un fragment frais pour la congélation, afin de pouvoir réaliser d’éventuelles études cytogénétiques (FISH, CGH array, RNA-Seq, etc.), même si certaines de ces techniques sont parfois désormais réalisables à partir d’un prélèvement fixé dans le formol.
Sarcomes
Les sarcomes ne sont pas rares chez le nouveau-né et le nourrisson, et peuvent être primitifs ou secondaires, métastatiques. Dans cette tranche d’âge, ce sont en général des sarcomes de haut grade : fibrosarcome infantile/congénital, tumeur rhabdoïde, rhabdomyosarcome(3).
Le fibrosarcome congénital est diagnostiqué dans les 2 premières années de vie. La lésion prédomine chez le garçon et survient préférentiellement sur les extrémités distales, moins souvent sur la tête ou le cou, sous forme d’une masse arrondie de croissance très rapide, vite volumineuse (> 10 cm), souvent congénitale ou survenant dans les semaines qui suivent la naissance. La peau en regard est tendue, brillante, parcourue de longues télangiectasies (figure 1). Dans quelques cas, la tumeur, très angiomateuse, s’ulcère largement simulant un hémangiome infantile de type RICH, d’autant qu’une coagulopathie peut être associée. Le diagnostic est confirmé par l’étude en FISH en raison d’une récurrence de la translocationt (12 ; 15) (p13 ; q26) avec gène de fusion ETV6-NTRK3, mais il existe quelques cas où cette translocation est absente. Dans tous les cas, le diagnostic moléculaire doit être demandé. Le traitement de choix est l’ablation chirurgicale large. Une polychimiothérapie préopératoire peut réduire la masse tumorale ou, en post-opératoire, compléter le geste chirugical. Le taux de guérison à 5 ans est supérieur à 90 % malgré un taux de récidive de 30 % environ.Les métastases sont exceptionnelles(4).
Le dermatofibrosarcome dans sa forme congénitale (DFS) est exceptionnel. Il peut être très trompeur cliniquement, car il peut mimer un angiome en touffes, un hamartome, une aplasie cutanée devant des plaques infiltrées et atrophiques (figure 2). Il est donc souvent diagnostiqué avec du retard(5).
Chez l’enfant plus grand, les sarcomes de bas grade prédominent dans la peau, mais on doit aussi penser à l’exceptionnel rhabdomyosarcome cutané primitif. Le sarcome le plus fréquent dans cette tranche d’âge est le dermatofibrosarcome. Il est caractérisé, comme chez l’adulte, par la fréquence élevée des récidives locales, sans métastase ganglionnaire ou viscérale. Entre 5 et 10 % de ces tumeurs apparaissent chez l’enfant prépubère. Il s’agit alors le plus souvent d’une plaque infiltrée, ferme à la palpation, rouge ou brune. Après quelque temps d’évolution, des nodules peuvent se développer, mais la lésion peut être longtemps asymptomatique. Elle peut devenir douloureuse (figure 3). Des formes multiples ont été décrites en association à un déficit en adénosine déaminase (ADA)(6). Une transformation en sarcome de haut grade est rare mais possible. La prise en charge est la même que chez l’adulte(7).
Le fibroblastome à cellules géantes (FCG) est considéré comme la forme juvénile du DFS, montrant les mêmes anomalies génétiques. Il se présente comme une masse dermo-hypodermique à développement lent, fixée à la peau, asymptomatique, et son évolution est identique à celle du DFS.
Le diagnostic est histologique, toujours confirmé par la recherche de la translocation t (17 ; 22) de fusion COL1A1/PDGF β, qui est quasi-constante (figure 4). La formation d’un anneau chromosomique surnuméraire dérivéde t (17 ; 22) est fréquente chez l'enfant(5).
L’évolution naturelle est très lente, sur des années voire des dizaines d’années. Le diagnostic chez l’enfant est souvent porté avec beaucoup de retard(5). Le traitement de choix est l’excision élargie, au mieux par chirurgie de Mohs (slow Mohs) (figure 5).
La tumeur fibrohistiocytaireplexiforme (TFHP) est également un sarcome de bas grade de malignité, presque exclusivement observé chez l’enfant et l’adolescent. Elle peut être ubiquitaire mais est le plus souvent située aux membres supérieurs. Cette tumeur se présente comme un nodule sous-cutané dur, asymptomatique ou douloureux, souvent de moins de 3 cm de diamètre. La tumeur peut infiltrer les plans musculaires profonds et être très mutilante. Le diagnostic est uniquement fondé sur l’histologie, car aucune anomalie génétique n’a été retrouvée à ce jour (figure 6). La TFHP est agressive localement et récidive fréquemment (40 %). Son traitement est l’exérèse chirurgicale large et complète(8).
L’histiocytofibrome angiomatoïde est un autre sarcome de bas grade observé quasi-exclusivement chez l’enfant et l’adolescent. Il est rare, de croissance lente, situé sur les extrémités ou le tronc (figure 7). Cliniquement, il s’agit d’une tumeur ou masse unique, située plus ou moins profondément dans le tissu sous-cutané, sans caractéristique clinique permettant de la reconnaître. Le diagnostic est histologique et moléculaire : translocation 16-12 (16p11-12q13) et fusion des gènes FUS et ATF1. L’évolution est relativement favorable avec moins de 10 % de récidives et moins de 1 % de métastases ganglionnaires. Le traitement de choix est l’exérèse complète large(9).
Le rhabdomyosarcome est l’une des tumeurs malignes les plus fréquentes de l’enfant. Une présentation cutanée primitive n’est pas exceptionnelle sous la forme d’une masse lobulée, infiltrée, adhérente à la peau, luisante, rosée à orangée ou pourpre, ferme au palper, finement télangiectasique, située surtout au niveau cervico-céphalique ou dans les régions génito-urinaires. Dans cette localisation, ces tumeurs sont exophytiques, en « chou-fleur », et peuvent être confondues avec des condylomes (sarcomes botryoïdes). Le type histologique le plus fréquent est le type embryonnaire, les types botryoïdes ou alvéolaires sont plus rares. Le traitement est lourd associant une excision large, une polychimiothérapie, et éventuellement une radiothérapie. Le pronostic semble meilleur si l’enfant est jeune. Le type histologique est essentiel pour le pronostic. Le rhabdomyosarcome alvéolaire a le risque le plus élevé d’expansion rapide et de métastases(10).
Quant à l’angiosarcome cutané, il est extrêmement rare chez l’enfant. Il siège préférentiellement aux membres inférieurs et survient souvent sur un terrain particulier : une malformation vasculaire, un lymphœdème ou une hémi-hypertrophie congénitaux, un hémangiome congénital traité par radiothérapie ou un xeroderma pigmentosum(11).
Parmi les tumeurs vasculaires malignes de l’enfant, le sarcome/maladie de Kaposi se voit au cours de l’infection VIH. Elle est devenue exceptionnelle chez l’enfant. Sa présentation clinique est comparable à celle que l’on connaît chez l’adulte.
Le sarcome d’Ewing est le troisième sarcome le plus fréquent de l’enfant et de l’adolescent. Son siège de prédilection est l’os, l’atteinte cutanée primitive est très rare. Dans une étude comparative des sarcomes d’Ewing primitifs de l’os et primitifs de la peau, on constate que les tumeurs cutanées se voient plus souvent chez la fille, plus souvent aux membres avec une taille tumorale moyenne de 2 à 3cm(12). Le diagnostic est histologique, nécessite une confirmation moléculaire de préférence sur du matériel congelé afin d’identifier une translocation spécifique du gène EWSR1 en FISH ou en RT-PCR. Le pronostic des formes cutanées primitives apparaît meilleur, peut-être lié à la taille plus petite des tumeurs lors du diagnostic, à un diagnostic plus précoce lié à la visibilité de la tumeur et au plus faible pourcentage de métastases lors du diagnostic. La survie globale à 10 ans est d’environ 90 %.
Moins de 2 % des tumeurs musculaires, qu’elles soient bénignes ou malignes, sont rapportées chez l'enfant(10). Le léiomyosarcome est donc exceptionnel. On le décrit dans le contexte d’une infection VIH avec coinfection par l’EBV. Ce dernier virus est mis en évidence par hybridation in situ au sein des cellules tumorales(13).
Hématodermies
Les hématodermies sont les tumeurs malignes les plus fréquentes chez le nouveau-né et le nourrisson, représentées en premier lieu par les atteintes cutanées des leucémies, essentiellement celles à composante monoblastique ou myélo/mono-blastique, parfois « aleucémiques » au moment du diagnostic, c’est-à-dire sans blastes circulants(3,14). Ce sont des urgences diagnostiques, et le dermatologue est souvent en première ligne. Chez l’enfant plus grand, le mycosis fongoïde (MF) prédomine, suivi des lymphoproliférations cutanées primitives CD30+ et des lymphomes/leucémies lymphoblastiques. Le MF est souvent diagnostiqué tardivement, car il se manifeste par des lésions hypochromiques ou mimant un pityriasis lichénoïde. Il n’y a pas de lymphome B centro-folliculaire, et le lymphome B de la zone marginale est très rare avant l’adolescence.
Les leucémies sont les tumeurs malignes les plus fréquentes de l’enfant, représentant plus de 30 % des cancers pédiatriques. L’atteinte cutanée est rare et se voit plus souvent au cours des leucémies aiguës myéloïdes (LAM)(3). L’aspect clinique diffère selon les tranches d’âge. Dans la période néonatale, le tableau clinique peut être celui d’un blueberry muffin baby (figure 8). Il s’agit d’une éruption présente à la naissance, faite de macules et de papules bleutées de moins de 1 cm, souvent disséminées. Les étiologies du blueberry muffin baby sont résumées dans le tableau 1. Les LAM congénitales sont le plus souvent de type 4 ou 5, avec une incidence élevée de translocations sur le chromosome 11 avec un point de cassure 11q23. Elles sont à différencier des syndromes myéloprolifératifs transitoires survenant typiquement au cours des trisomies 21. Les lymphomes sont très rares chez l’enfant, le moins rare étant représenté par le mycosis fongoïde comme chez l’adulte.
Le mycosis fongoïde (MF) touche le grand enfant et l’adolescent avec des plaques érythémato-squameuses fixes discrètement infiltrées, prises souvent à tort pour de l’eczéma ou du psoriasis. Chez l’enfant, il n’est pas rare que le MF se manifeste sous la forme de plaques hypopigmentées (figure 9).
Le diagnostic histologique est souvent difficile car ces MF, contrairement à ceux de l’adulte, sont plus souvent de phénotype CD8 comme dans la plupart des lésions inflammatoires. La recherche d’une clonalité T peut être utile mais un clone T peut aussi se rencontrer dans d’autres entités comme le pityriasis lichénoïde(15). L’évolution est la même que chez l’adulte, chronique, et une transformation en lymphome de haut grade est possible (figure 10). Le traitement n’est pas différent de celui de l’adulte.
• Les lymphoproliférations cutanées CD30+ primitives sont surtout représentées par la papulose lymphomatoïde. Le lymphome primitif CD30+est beaucoup plus rare. L’âge moyen d’apparition est de 7 ans, mais elles peuvent se voir chez l’enfant très jeune. Elles s’accompagnent fréquemment d’un prurit et surviennent sur un terrain atopique dans près de 30 % des cas. Elles sont dans plus d’un tiers des cas associées à un pityriasis lichénoïde et sont souvent déclenchées/précédées par un épisode infectieux. Elles laissent très souvent des cicatrices dont il faut tenir compte dans la prise en charge thérapeutique si les poussées sont fréquentes et multilésionnelles (figure 11). Les lésions sont papulo-nodulaires et parfois nécrotiques. L’histologie met en évidence un infiltrat inflammatoire mixte dans le derme, d’architecture triangulaire à pointe en bas, dans lequel s’observent de grandes cellules CD30+.
Chez l’enfant, l’infiltrat est particulièrement riche en éosinophiles(16). Le diagnostic est toujours fondé sur la confrontation anatomoclinique, car l’histologie, à moins que les grandes cellules atypiques soient nettement majoritaires, n’est pas très spécifique et peut faire discuter des lésions de prurigo/piqûres d’insectes/nodules post-scabieux. En effet, ces lésions sont très fréquentes chez l’enfant et histologiquement elles peuvent aussi contenir des cellules atypiques CD30+. L’autre diagnostic différentiel parfois malaisé est celui de pityriasis lichénoïde, qui peut être également très nécrotique et contenir des cellules CD30+. Les lésions sont en général plus nombreuses(15). Lapapulose lymphomatoïde évolue par poussées successives involuant spontanément. Le risque de survenue possible à l’âge adulte d’un deuxième lymphome nécessite une longue surveillance(16).
Le lymphome lymphoblastique cutané, le plus souvent de phénotype B, révèle souvent une leucémie aiguë lymphoblastique. Il se présente, en général, sous la forme d’un ou de plusieurs nodules tendus, fermes, rouges vifs, situés sur la tête et le cou (figure 12). L’histologie et l’immunohistochimie retrouvent une prolifération clonale de lymphoblastes immatures B.
Ce lymphome extrêmement agressif nécessite un traitement en urgence, car il est d’autant plus efficace qu’il est commencé tôt. Il s’agit d’un lymphome très chimio-sensible. Le lymphome lymphoblastique cutané T est plus rare(14,17).
Enfin, le lymphome à typed’hydroa vacciniforme est exceptionnel dans nos contrées. Il se rencontre surtout en Amérique latine et dans les pays asiatiques (Japon, Corée, Taïwan). C’est un lymphome T cytotoxique (CD3, CD8) angiocentrique associé à l’EBV et classé dans le groupe des « syndromes lymphoprolifératifs associés à l’EBV » dans la dernière version de la WHO-EORTC classification(18).
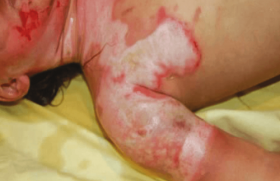
L’histiocytose langerhansienne est une prolifération clonale d’histiocytes, avec des formes limitées à la peau ou multisystémiques. Les formes néonatales (figure 13) sont en général limitées à la peau et sont le plus souvent d’involution spontanée en quelques semaines (histiocytoses auto-involutives d’Hashimoto-Pritzker) alors que les formes d’apparition plus tardives sont souvent accompagnées d’une atteinte systémique multi-organes. En période néonatale, elles peuvent se présenter sous la forme d’un blueberry muffin rash où la présence de petits nodules croûteux noirâtres peut orienter le diagnostic. Ce peut être aussi une éruption bulleuse érosive et hémorragique faisant croire, à tort, à une infection herpétique.
Plus tard, l’atteinte dermatologique la plus classique est une éruption profuse de macules et de papules squameuses, jaunâtres, de petites tailles associées à des vésicules et du purpura. L’atteinte du tronc en maillot de corps est typique, mais tout letégument peut aussi être atteint, y compris le cuir chevelu, les ongles, les paumes, les plantes et les muqueuses. Les lésions sont parfois moins typiques, plus frustes, mises en évidence par un examen dermatologique attentif. Quelle que soit la présentation cutanée, une surveillance prolongée est nécessaire(19,20). Le diagnostic doit toujours être confirmé par la biopsie qui montre un infiltrat dermique épidermotrope de cellules de taille moyenne à cytoplasme éosinophile à noyau excentré et réniforme, exprimant le CD1a et le CD207 (Langerine). La recherche de mutations BRAF ou d’une autre mutation dans la voie des MAP kinases fait aujourd’hui partie du bilan systématique de ces histiocytoses permettant un éventuel traitement ciblé(20).
Neuroblastome métastatique
Le neuroblastome est l’un des cancers les plus fréquents de la première année de vie. Il se manifeste au niveau de la peau par des métastases sous-cutanées qui se voient dans la période néonatale dans un tiers des cas, révélant en général le diagnostic(3). On peut y penser devant des papulo-nodules fermes, indolores, mobiles, de couleur bleutée ou violacée, entourés d’un halo de vasoconstriction. La présentation clinique peut être celle d’un blueberry muffin baby. Le diagnostic est histologique. En dépit du caractère métastatique, le pronostic est plutôt bon chez le nouveau-né, la tumeur pouvant régresser spontanément ou se transformer en tumeur bénigne (ganglioneurome).
Mélanome pédiatrique
Le mélanome de l’enfant prépubère est très rare. Il en existe deux grandes variétés : le mélanome associé au nævus congénital de grande taille et le mélanome de Spitz. Ce dernier n’est associé à aucun facteur de risque. Il peut être difficile à diagnostiquer cliniquement, pouvant mimer avant tout un nævus de Spitz, un granulome pyogénique, un xanthogranulome. On doit s’inquiéter devant tout nævus de Spitz de grande taille (> 1 cm), à fortiori ulcéré et hémorragique.
À partir de la puberté, on voit apparaître des mélanomes conventionnels de type adulte (SSM). Les mélanomes associés aux nævus congénitaux se développent tôt dans la vie (1-5 ans), chez des enfants ayant un nævus congénital de très grande taille, d’autant plus que les lésions sont multiples (≥ 2), avec atteinte du système nerveux central(21).
Les tumeurs malignes de l’enfant sont rares, et leur diagnostic clinique est difficile, réalisant souvent un tableau de blueberry muffin baby dans la période néonatale ou pouvant prendre l’aspect d’une tumeur bénigne ou d’une dermatose inflammatoire. La biopsie cutanée, indispensable au diagnostic, doit être réalisée rapidement afin d’établir le diagnostic le plus précoce possible. S’il n’y a pas toujours d’urgence à faire une biopsie, le retard au diagnostic est dans bien des cas préjudiciable à une prise en charge pluridisciplinaire et donc au pronostic.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :