Publié le 23 avr 2009Lecture 6 min
Le déficit en prolidase : rare et difficile à prendre en charge. À propos d’un cas
N. ELKIHAL, K. SENOUCI, B. HASSAM Service de dermatologie, hôpital Ibn-Sina, Rabat (Maroc)
Le déficit en prolidase est une maladie autosomique récessive rare. Sa prévalence dans le monde est inconnue. Quelque 40 cas ont été rapportés. Elle se traduit dès l’enfance par des ulcères des jambes et des pieds en rapport avec un trouble de cicatrisation. Nous rapportons l’observation d’une patiente suivie depuis l’âge de 1 an dans notre service.
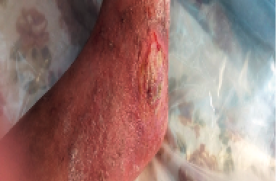
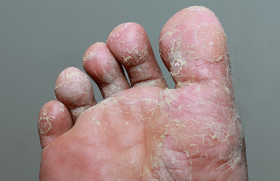
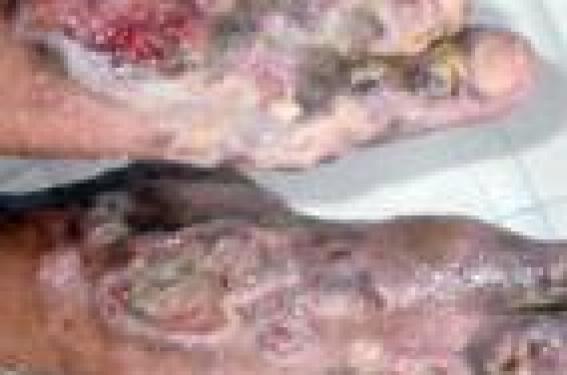
E.H., âgée de 16 ans, est suivie dans notre service depuis l’âge de 1 an pour des ulcères récidivants des pieds. Enfant adoptée dès la naissance, nous ne disposons d’aucune information sur ses antécédents, ni de renseignements sur ses parents et sa fratrie. Elle est bien vaccinée selon le PNI et est scolarisée dès l’âge de 7 ans. Figure 1. Ulcères bilatéraux du dos des pieds. • La patiente a présenté depuis le début de la marche un ulcère du dos du pied gauche, douloureux et prurigineux, qui a persisté alors que d’autres ulcères bilatéraux de taille variable sont apparus. À l’examen, le fond de l’ulcère est couvert de fibrine ; lorsqu’il est détergé, il a l’aspect bourgeonnant, mais il ne cicatrise pas. La zone péri-ulcéreuse est épaissie avec pachyépidermie, dépigmentation et hyperpilosité paradoxale. Les pouls périphériques sont présents. Le reste de l’examen est normal en dehors d’une peau sèche et fine, d’une arachnodactylie et de télangiectasies au niveau du dos des deux mains. • La NFS a mis en évidence une anémie (Hb : 6,4 g/100 ml) hypochrome microcytaire ferriprive. La VS est à 75 mm. Il n’y a pas d’hémolyse. La radiographie des pieds et le Doppler artériel et veineux des deux membres inférieurs étaient normaux. L’échographie abdominale n’a pas objectivé d’hépatosplénomégalie. • Une biopsie a été réalisée avec un résultat en faveur d’un lichen striatus. • Le traitement a consisté en des soins locaux à base d’éosine, des dermocorticoïdes et des antibiotiques administrés localement et par voie orale, mais n’a pas apporté d’amélioration ; elle reçoit par ailleurs du fer pour corriger son anémie. • Un dosage quantitatif des acides aminés urinaires est réalisé, objectivant une importante iminodipeptidurie faisant évoquer un déficit en prolidase. • L’examen lors de la dernière hospitalisation montre de multiples ulcères au niveau des deux pieds, superficiels, localisés aux dos et aux plantes des pieds et au niveau des inter-orteils (figures 1 et 2). Les ulcères ont un fond fibrineux, les bords sont irréguliers avec une peau péri-ulcéreuse scléreuse et une zone d’atrophie blanche. • L’évolution est marquée par la persistance des ulcères, l’incurvation des pieds handicapant la marche ; l’anémie a disparu (Hb à 12,9 g/100 ml). La radiographie des pieds met en évidence une déminéralisation osseuse diffuse (figure 3). La biopsie de la graisse abdominale à la recherche d’une amylose associée se révèle négative ainsi que la recherche d’une protéinurie. Figure 2. Ulcères plantaires. • Le traitement symptomatique des ulcères, l’administration de vitamine C et de Pyostacine® en cas d’infection n’apportent aucune amélioration. La greffe cutanée a été envisagée avec les plasticiens, mais les données de la littérature n’étaient pas encourageantes. Discussion Le déficit en prolidase est une maladie autosomique récessive très rare. Ce déficit a été mis en évidence pour la première fois en 1974 sur les érythrocytes et les leucocytes et, en 1977 et 1979, sur les fibroblastes dermiques. La prolidase (peptidase D, imidopeptidase) est une enzyme qui hydrolyse la proline ou l’hydroxyproline à l’extrémité C. Son gène est situé sur le chromosome 19p13.2. Elle intervient à la phase terminale du catabolisme du collagène. En conséquence, son déficit est responsable en amont de l’accumulation de dipeptides qui passent dans le sang, puis dans les urines ; en aval, la perte de dipeptides, dont les acides aminés ne sont pas réincorporés in situ, entraîne un déficit de la synthèse de procollagène, et donc une maladie de cicatrisation (figure 4). L’histologie montre une atrophie épidermique avec destruction irrégulière de la membrane basale ; au niveau des ulcères, on peut observer des dépôts de substance amyloïde sur la paroi interne des vaisseaux. Clinique. La maladie se traduit cliniquement par l’apparition chez un enfant d’ulcères des jambes et des pieds, invétérés, bilatéraux, résistant aux traitements. Le fond ulcéreux, lorsqu’il est détergé, est caverneux ou vasculaire et bourgeonnant, mais ne cicatrise pas. La zone péri-ulcéreuse est le siège d’une pachyépidermie avec dépigmentation et hyperpilosité paradoxale. Des troubles trophiques cutanés peuvent se voir, à type d’éruption inflammatoire, de Figure 3. Radiographie des pieds face (A) et profil (B). télangiectasies et de scléro-atrophies (tableau). D’autres signes sont associés au déficit en prolidase : syndrome dystrophique, sensibilité aux infections, complications ORL et ophtalmiques, hépatosplénomégalie, retard mental et psychomoteur plus ou moins sévère... Le diagnostic repose sur le dosage enzymatique érythrocytaire et le taux d’aminopeptides dans les urines. Le diagnostic anténatal est possible sur les cellules amniotiques. Chez notre patiente, le syndrome dystrophique est présent sous la forme d’une arachnodactylie. Le diagnostic est évoqué cliniquement par des ulcères des jambes et des pieds, bilatéraux, résistant aux traitements, survenant chez un enfant. Des cas familiaux peuvent exister, mais le défaut d’anamnèse ne permet pas de l’affirmer. Le diagnostic est confirmé biologiquement par l’iminodipeptidurie, qui est massive et constante. Le dipeptide prédominant est le glycylproline. Traitement • Le traitement symptomatique est celui de tous les ulcères : détersion, désinfection et application de pansements colloïdes. Pour certains auteurs, l’antibiothérapie doit être systématique ; nous pensons qu’elle est de mise en cas d’infection. • Le traitement étiologique consiste en la supplémentation en proline et hydroxyproline par voie locale, et en cofacteurs de la prolidase, la vitamine C et le manganèse. Ce traitement n’a pas été efficace chez notre patiente. Figure 4. Schéma du rôle de la prolidase dans le métabolisme du collagène. • La dapsone à dose de 75 mg/j pendant 2 mois a permis d’obtenir une épidermisation dans certaines observations ; ce traitement n’a pas été essayé chez notre patiente en raison de son anémie ferriprive. Des greffes de résilles ont été réalisées et se sont avérées efficaces au début de la maladie, mais le résultat n’a été que transitoire et des amputations ont dû être pratiquées. Conclusion Le déficit en prolidase est une maladie autosomique rare qui touche l’enfant. Il faut penser à cette pathologie rare devant des ulcères invétérés apparaissant dès l’enfance. Le diagnostic est posé devant une iminodipeptidurie positive, alors que le reste du bilan est normal. Le traitement est symptomatique mais décevant ; seul un traitement étiologique efficace pourra apporter un espoir à l’avenir.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :