Publié le 11 fév 2022Lecture 5 min
Syndrome de Werner : une observation inhabituelle avec préjudice esthétique d’évolution rapidement handicapante
Omar BOUDGHENE-STAMBOULI, CHU de Tlemcen (Algérie)*
Le syndrome de Werner est une génodermatose rare de transmission autosomique récessive liée dans près de 80 % des cas à une mutation du gène WRN. Il se manifeste en règle générale précocement dans la vie. Il est caractérisé par une atrophie pseudo-sclérodermique cutanée et musculaire, une ankylose articulaire, une déformation des pieds, des calcinoses et des ulcérations spontanées pouvant être invalidantes.
Nous rapportons un cas de syndrome de Werner exceptionnel avec un préjudice esthétique majeur, d’évolution rapidement très invalidante, confinant la malade au fauteuil roulant.
Observation
Une patiente âgée de 26 ans, issue d’un mariage consanguin de premier degré, présente une petite taille liée à un arrêt précoce de la croissance. Le début de la maladie semble remonter à l’âge de 7 ans, se manifestant par des calcinoses sous-cutanées, un faciès sclérodermiforme, un acrosyndrome (cyanose des pieds aggravée par le froid), une déformation en griffe des doigts, une peau difficile à plisser au niveau des avant-bras et jambes.
Lors de la consultation, elle montre un visage amaigri, émacié, avec un nez effilé saillant, des lèvres amincies, un menton étroit (figure 1). L’aspect du visage est très évocateur d’une « tête d’oiseau ». Une protrusion oculaire est notée sans atrésie de l’orifice buccal.
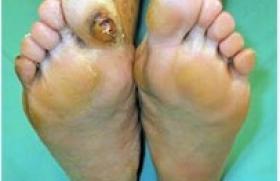
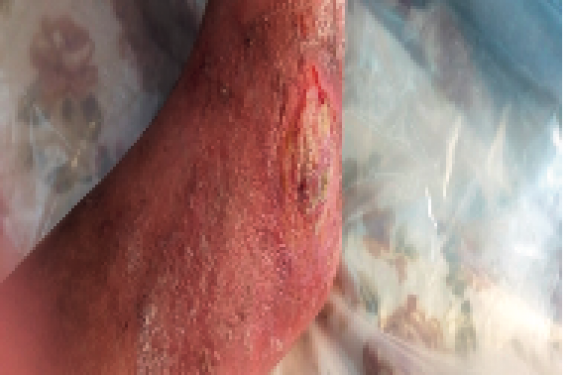
La patiente a des ulcérations traînantes périmalléolaires (figure 2) n’ayant aucune tendance spontanée à la cicatrisation, s’étendant et s’allongeant sur plusieurs centimètres, des calcinoses en regard de grosses articulations (figure 3), une déformation des pieds, une déformation en griffe des orteils (figure 4), une ankylose des coudes et des genoux, des ongles dystrophiques, des dents déchaussées avec de nombreuses caries. La canitie est présente, apparue vers l’âge de 15 ans ; l’alopécie diffuse mais incomplète.
Une lipodystrophie avec perte du tissu graisseux sous-cutané, prédominant au niveau des membres, est associée à une atrophie musculaire globale donnant un aspect maigre, voire cachectique, aux membres (aspect d’un cadavre congelé). Du fait d’une baisse de l’acuité visuelle, un examen ophtalmologique a été réalisé et pose un diagnostic de cataracte bilatérale précoce.
Le bilan radiologique met en évidence : une subluxation et une déformation des articulations méta-tarso-phalangiennes avec ostéoporose diffuse (figures 4 et 5) ; une atrophie des tissus musculaires et de la graisse sous-cutanée ; des déformations articulaires avec calcification des tissus mous.
Le bilan à la recherche d’une sclérodermie systémique est négatif du fait de la normalité des épreuves fonctionnelles respiratoires, de la manométrie œsophagienne ou du radio-cinéma œsophagien, de l’absence d’anticorps antinucléaires, d’anticorps centromères et d’anti-Scl-70.
Discussion
Le syndrome de Werner, syndrome de sénescence précoce, associe une petite taille, des anomalies cutanéo-phanériennes (canitie précoce, alopécie, dépilation, sclérose cutanée), des complications orthopédiques (pieds plats, hallux valgus et autres déformations articulaires) ainsi que des manifestations systémiques (cataracte précoce, athérosclérose prématurée et diffuse, endocrinopathies) et un risque élevé de certains cancers (sarcomes, hémopathie myéloïde).
Le décès survient vers 40-50 ans, généralement secondaire à un accident cardiovasculaire ou au développement d’une tumeur maligne.
Il s’agit d’une maladie rare, dont l’incidence est estimée de 1 à 22 cas par million d’habitants(1). Le Japon est le pays avec la plus forte incidence : près de 1 cas sur 300 000 habitants(2) et environ 80 % des 1 300 cas publiés en 2003(3). 70 % des patients sont nés de mariages consanguins au premier degré(4), et des antécédents de syndrome de Werner sont trouvés dans la fratrie pour 50 % des cas(3). Dans les pays du Maghreb, six cas ont été rapportés en Tunisie(5) dans une étude rétrospective descriptive du service de dermatologie du CHU Farhat-Hached à Sousse, colligeant sur une période de trente ans (1983– 2012) tous les patients pour lesquels le diagnostic du syndrome de Werner était confirmé selon les critères établis par The Seattle International Registry of Werner Syndrome.
Un cas en Algérie a été rapporté en 2011(6) chez une patiente avec une intolérance au glucose (HGPO), une insulino-résistance (taux de peptide C élevé), une consanguinité, un morphotype particulier, une cataracte bilatérale, une ostéoporose et une néoplasie thyroïdienne. La transmission est autosomique récessive(7), liée à une mutation du gène WRN, située sur le bras court du chromosome 8, codant pour la protéine WRN de la famille RecQ des ADN hélicases(8,9,10) (RecQ3).
Des mutations du gène LMNA, codant pour les lamines A et C, ont été mises en évidence chez une minorité de patients ayant un gène WRN intact. Ces patients avaient une forme plus sévère de la maladie et un début plus précoce(11) comme dans l’observation présentée. L’apparition des différents symptômes du syndrome de Werner se fait habituellement dans l’ordre suivant :
arrêt de croissance (à partir de la puberté),
canitie et altération de la voix (à l’adolescence),
modifications cutanées et cataracte (30 à 35 ans),
artériopathie (40 à 45 ans).
Dans notre observation, les modifications cutanées et la cataracte étaient beaucoup plus précoces. Il existe des formes frustes et des formes incomplètes.
Habituellement, l’affection s’aggrave lentement, entraînant un préjudice esthétique et des troubles fonctionnels notables. Dans le cas que nous avons présenté, l’affection a évolué vers un préjudice esthétique majeur très affichant, à évolution rapide très handicapante, confinant la malade au fauteuil roulant. Ce syndrome est associé à un risque élevé de cancer qui représente la première cause de décès chez les patients japonais(3), d’où la nécessité d’une surveillance étroite des malades afin de dépister toute néoplasie débutante.
Le diagnostic différentiel peut se faire avec deux groupes d’affections : les maladies avec sclérose diffuse, notamment la sclérodermie systémique et les morphées monoméliques, et les atrophies cutanées congénitales ou familiales débutant dans les premiers mois ou années de vie (acrogeria, syndrome de Cockayne, syndrome de Rothmund-Thomson...). L’élévation de l’acide hyaluronique urinaire est un bon marqueur diagnostique biologique du syndrome de Werner(12). Des critères du diagnostic clinique de syndrome de Werner ont été établis(13). Une analyse génétique moléculaire peut actuellement être réalisée à la recherche d’une mutation homozygote du gène WRN(8) .
Dans la plupart des cas, il s’agit de mutations bialléliques entraînant un décalage du cadre de lecture ou l’apparition d’un codon-stop(8).
Conclusion
Tout signe clinique évocateur d’une sénescence prématurée, qu’elle soit cutanée comme un grisonnement prématuré des cheveux, un état sclérodermiforme et « vieilli » des extrémités, ou viscérale comme une cataracte précoce doit faire évoquer le diagnostic de syndrome de Werner. La reconnaissance précoce de cette affection doit permettre la mise en place d’un suivi adapté ainsi que le dépistage et le traitement des complications cardiovasculaires, oculaires, endocriniennes et tumorales.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :