Publié le 19 aoû 2008Lecture 6 min
Des tibialgies rebelles
G. AYOUB, J.-D. LAREDO, Hôpital Lariboisière, Paris
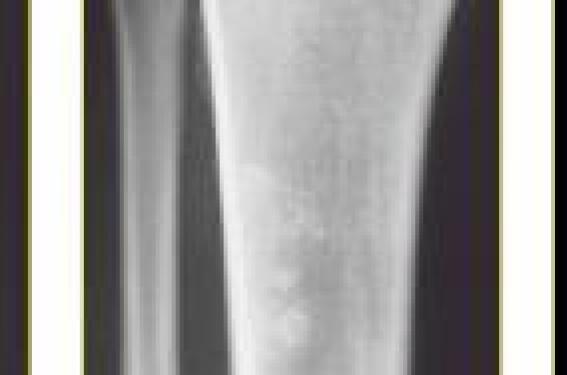
Un homme de 53 ans consulte pour des douleurs des tibias, prédominant du côté droit.
Ces douleurs, d’apparition insidieuse, évoluent depuis plusieurs semaines. Elles sont d’horaire mixte, et sont incomplètement soulagées par les antalgiques. Elles surviennent cependant sur un contexte particulier, fait d’urticaire chronique, d’inflammation biologique, et d’hypergammaglobulinémie monoclonale IgM kappa. Les radiographies standards montrent des lésions condensantes de l’extrémité supérieure de la diaphyse tibiale droite (figures 1 et 2). Figure 1. Radiographie standard montrant plusieurs petites images de condensation modulaire de l’extrémité supérieure de la diaphyse tibiale droite. Figure 2. Agrandissement sur la diaphyse tibiale droite montrant plus nettement la présence de plusieurs lésions condensantes bien limitées. Figure 3. IRM T1 coupe coronale montrant que les lésions vues en radiographie sont plutôt confluentes, en hyposignal T1. En IRM, ces lésions sont en hyposignal T1, rehaussées par l’injection de gadolinium, et en hypersignal STIR (figures 3 à 7). Figure 4. IRM STIR coupe coronale montrant les lésions osseuses en hypersignal. Figure 5. IRM T1 Gado coupe coronale montrant le rehaussement important des lésions. Figure 6. IRM T1 Gado avec zoom sur les lésions, rehaussées après injection de gadolinium. Figure 7. IRM T1 Gado coupe axiale montrant les lésions de la diaphyse tibiale droite rehaussées par le gadolinium. Quel est votre diagnostic ? L’ensemble de ces données, et notamment le contexte de survenue, fait retenir le diagnostic de syndrome de Schnitzler. Commentaires Le syndrome de Schnitzler a été décrit pour la première fois dans les années 70 par le dermatologue qui lui a donné son nom. Le diagnostic repose sur la présence d’une gammapathie monoclonale IgM et d’un rash cutané chronique, associés à deux critères parmi les suivants : fièvre intermittente, hépato- ou splénomégalie, arthralgies ou arthrites, douleurs osseuses, hyperleucocytose et VS élevée. La maladie est rare, avec près d’une soixantaine de cas décrits, la majorité en Europe. Elle survient autant chez les hommes que chez les femmes, avec des âges extrêmes allant de 29 à 77 ans. Une errance diagnostique est souvent rapportée dans les cas publiés. La gammapathie monoclonale IgM est le plus souvent à chaîne légère kappa (près de 90 % des cas). Le taux d’IgM est supérieur aux valeurs normales (0,6-2,5 g/l) et durant l’évolution il peut soit rester stable soit continuer à augmenter. Une protéinurie de Bence-Jones peut se rencontrer dans plus de 40 % des cas. Une maladie de Waldenström est à éliminer si les taux d’IgM sont très élevés. Les biopsies de moelle sont normales dans 80 % des cas. Ailleurs, elles peuventmontrer une infiltration non spécifique, polyclonale, lymphocytaire et plasmocytaire. Le rash cutané chronique est monomorphe, parfois confluent. Il est essentiellement fait de papules érythémateuses, habituellement non prurigineuses. Elles sont sur le tronc et les membres, mais épargnent la tête et le cou. Elles évoluent par poussées, parfois entrecoupées d’un intervalle libre pouvant aller jusqu’à 2 semaines. De nouvelles lésions apparaissent tous les jours, et elles durent moins de 24 h. Des cas où l’apparition des lésions faisait suite à une ingestion d’alcool, ont été rapportés. L’histologie montre un infiltrat neutrophilique. Le traitement vient difficilement à bout des lésions cutanées. Il fait souvent appel aux antihistaminiques, aux corticoïdes, à la disulone, à la colchicine, aux AINS, voire aux immunosuppresseurs. Une fièvre est notée dans 90 % des cas. Elle est typiquement intermittente, avec des pics à 40°C, le plus souvent bien tolérés. La survenue des pics fébriles est indépendante du rash cutané. Des adénopathies se rencontrent dans la moitié des cas. Une hépato- ou une splénomégalie sont notées chez un patient sur trois. Une atteinte musculosquelettique survient chez près de 80 % des patients, et peut prendre la forme de douleurs osseuses, de myalgies ou d’arthralgies auxquelles s’associent parfois de véritables arthrites. Les douleurs osseuses, qui touchent à elles seules près de 70 % des patients, sont essentiellement localisées au pelvis et aux fémurs, et moins fréquemment, aux tibias, au rachis, aux clavicules et aux avant-bras. Les radiographies standards montrent typiquement des lésions condensantes, même si des lésions lytiques ont déjà été rapportées. Des réactions périostées sont possibles. La scintigraphie osseuse peut montrer une hyperfixation, correspondant à une ostéosclérose, dans près de 40 % des cas. L’IRM montre une infiltration médullaire mal limitée, en hypoT1 et hyperT2. Plusieurs diagnostics différentiels peuvent se discuter devant un syndrome de Schnitzler dont : • une maladie de Still de l’adulte, où les éléments distinctifs sont l’absence de gammapathie monoclonale IgM et la présence de taux élevés de ferritinémie ; • une hémopathie à type de lymphome ou de maladie de Waldenström. Dans ces cas, une biopsie d’une adénopathie ou une biopsie de moëlle osseuse permettent de corriger le diagnostic ; • une cryoglobulinémie où les signes cliniques sont température- dépendants et s’accompagnent d’une cryoglobulinémie sérique ; • un lupus érythémateux disséminé où l’on retrouve les signes cliniques évocateurs du lupus ainsi que des anticorps antinucléaires ; • un syndrome CINCA qui débute dans l’enfance et se caractérise par une atteinte neurologique et des déformations articulaires ; • un syndrome de Muckle-Wells marqué par la présence d’une histoire familiale évocatrice, d’une surdité et d’une amylose, et l’absence de composante monoclonale ; • un syndrome d’hyper-IgD caractérisé par une augmentation du taux d’IgD. L’évolution du syndrome de Schnitzler est marquée par l’absence de rémission spontanée. Le traitement est symptomatique et peu efficace. Un désordre lymphoprolifératif survient au bout de 10 à 20 ans dans près de 15 % des cas. Il s’agit le plus souvent d’un lymphome, d’un myélome, ou d’une maladie de Waldenström. Une amylose AA peut également venir compliquer la maladie. Quelques cas cliniques ont rapporté des améliorations après traitement par rituximab ou ciclosporine, mais de plus en plus de publications font mention de l’efficacité, parfois spectaculaire, de l’anakinra, antagoniste du récepteur de l’IL- 1, dans des cas de syndrome de Schnitzler parfois réfractaire. Dans une étude publiée en 2007, 11 patients ont été traités par péfloxacine avec des résultats encourageants.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :



