Publié le 11 fév 2010Lecture 4 min
Une lésion ulcéro-végétante du pied
R. CHRAÏBI, Y. AFIFI, H. CHRAÏBI, K. SENOUCI, B. HASSAM, Service de dermatologie, CHU Ibn Sina, Rabat, Maroc
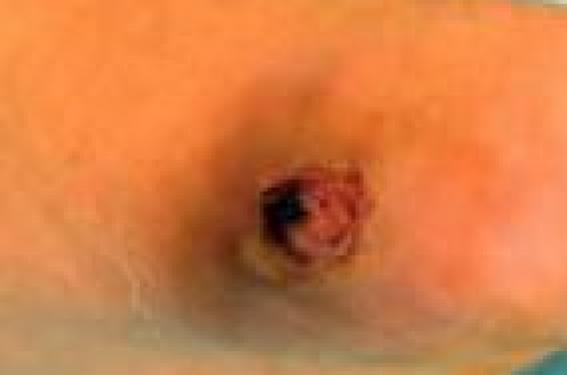
Monsieur B.E., âgé de 21 ans, sans antécédents pathologiques particuliers, est admis dans le service de dermatologie pour une lésion ulcéro-végétante du pied gauche.
Celle-ci a débuté il y a 4 ans par l’installation d’une papule érythémateuse indolore qui a progressivement augmenté de taille jusqu’à devenir ulcérée puis bourgeonnante. À son admission, le patient était en bon état général, présentant au niveau du bord antéro-interne du pied gauche, un nodule ferme, de couleur chair de 3 cm/2 cm, à surface érodée hétérogène et à base infiltrée entourée d’un îlot d’hyperkératose. La peau périlésionnelle était le siège d’un placard brunâtre infiltré (figure 1). Le reste de l’examen somatique était sans particularités. Une biopsie cutanée a été réalisée (figure 2). Figure 1. Lésion infiltrée de la face interne du pied gauche. Figure 2. biopsie cutanée. Diagnostic L’étude histologique a objectivé une prolifération irrégulière de cellules fusiformes associées à des cellules épithéliales. Un immunomarquage est réalisé, montrant une posivité pour l’anti-vimentine. La PS100 et la kératine étaient négatives. Sur les données de l’examen histologique et l’immunomarquage, le diagnostic de synovialosarcome biphasique grade II est prononcé. Un bilan d’extension a été réalisé comprenant radiographie du pied, échographie abdominale, tomodensitométrie thoracique et pelvienne : ces examens étaient normaux. En revanche, l’imagerie par résonance magnétique du pied a objectivé un processus lésionnel péri-métatarsien prédominant sur le bord interne du pied gauche. Une amputation du premier rayon du pied a été réalisée. Un traitement adjuvant comprenant 6 cures d’adriamycine a été instauré. Le recul est de 36 mois sans récidive locale ni métastases à distance. Commentaire • Le synovialosarcome représente 7 à 10 % de l’ensemble des sarcomes des tissus mous. C’est une tumeur de l’adulte jeune. L’âge moyen de survenue se situe entre 25 et 34 ans(1) avec une légère prédominance masculine (sex ratio : 1,5) (2). Notre patient était donc un cas relativement précoce. • Cette tumeur se présente le plus souvent sous forme nodulaire, évoluant de façon insidieuse, ce qui explique le retard fréquent au diagnostic. Dans notre observation, l’atypie et la latence clinique ont été responsables d’un retard diagnostique de 4 ans. • Sur le plan histologique, le déplacement au sein de la classification OMS des synovialosarcomes dans le groupe des tumeurs à histogenèse inconnue ou controversée, rend compte de l’incertitude sur l’origine de ces tumeurs (3). Le synovialosarcome se caractérise par la coexistence de plusieurs aspects histologiques (4). Il associe, dans une proportion variable, des éléments d’aspect épithélial, rappelant ceux des carcinomes, et des cellules fusiformes d’aspect fibrosarcomateux. La présence de l’un ou des deux contingents détermine le type mono- ou biphasique. • Le synovialosarcome dans sa forme biphasique présente peu de diagnostics différentiels et ne nécessite pas en général d’étude immunohistochimique, qui ne fait que confirmer le diagnostic (5). Dans notre observation, le diagnostic n’a été retenu qu’après de nombreuses relectures des lames. Le principal diagnostic différentiel était le sarcome épithélioïde. L’ensemble des aspects morphologiques, ainsi que la présentation clinique, permettent en général d’éliminer certains diagnostics tels qu’un sarcome épithélioïde (6), un sarcome à cellules claires, un schwannome malin, un carcinosarcome… Dans les cas difficiles, le diagnostic peut s’obtenir par la mise en évidence de translocations t(X,18) (p11.2;q11.2), qui sont spécifiques et retrouvées dans 70 % des synovialosarcomes (5). L’étude génétique était normale chez notre patient. • Vu le grand potentiel de récurrences locales et de métastases de ces tumeurs, un traitement agressif est impératif, incluant la résection chirurgicale en monobloc voire l’amputation. La radiothérapie complémentaire est largement indiquée dans les synovialosarcomes des extrémités. Des séries rétrospectives mentionnent l’intérêt de la chimiothérapie par adriamycine ou isofosfamide (7). Chez notre patient, on n’a pas eu recours à la radiothérapie, considérant que le geste chirurgical a été large. L’adjuvant thérapeutique a été la chimiothérapie. • L’évolution de ces tumeurs est dominée par la survenue de métastases pulmonaires et osseuses (10 à 20 % des cas). L’atteinte ganglionnaire est notée dans 6 à 27 % des cas et les récidives locales dans 25 à 35 % des cas. La première diffusion métastatique se fait en règle dans la première année d’évolution (7). Dans notre observation, le recul après traitement est de 36 mois, sans récidive ni locale ni à distance.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :



