Maladie de système, Médecine interne
Publié le 21 sep 2018Lecture 6 min
Syndrome d’Ehlers-Danlos : entre risques de sur- et sous-diagnostic
Karelle BENISTAN, Hôpital Raymond Poincaré, Garches
Les syndromes d’Ehlers-Danlos (SED) sont un groupe hétérogène de maladies héréditaires du tissu conjonctif caractérisées par une hyperlaxité articulaire et cutanée, et une fragilité des tissus conjonctifs, dues à des anomalies de la biosynthèse et/ou de la structure des protéines de la matrice extracellulaire. Leur transmission est le plus souvent autosomique dominante. Leur prévalence est estimée à 1 pour 5 000. Plusieurs classifications se sont succédées. La classification de Villefranche proposée en 1997, a été utilisée jusqu’en 2017, reconnaissant 6 types (sur des critères génétiques et cliniques) : classique, hypermobile, vasculaire, cyphoscoliotique, arthrochalasique et dermatosparaxis. Cette classification ne paraît plus adaptée depuis 1997 en raison de la découverte d’autres gènes impliqués dans les SED.
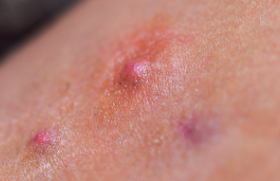
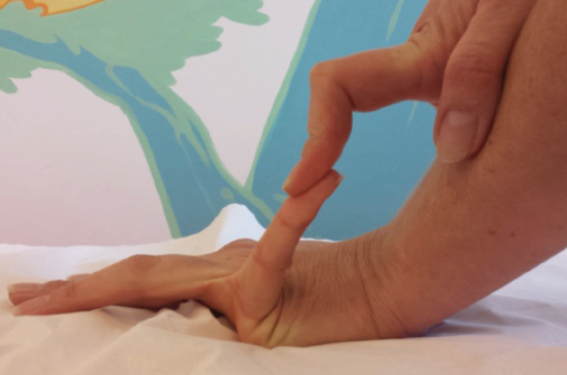
• Le syndrome d’Ehlers-Danlos hypermobile est le plus fréquent. Il semble se manifester davantage chez les femmes que chez les hommes et être de transmission autosomique dominante. Ses bases moléculaires ne sont actuellement pas encore élucidées. Il se manifeste par une hyperlaxité articulaire généralisée, des entorses et des luxations à répétition, avec un tableau douloureux chronique retentissant sur la qualité de vie (figure 1). L’hyperlaxité articulaire se mesure au moyen du score de Beighton (tableau) (figures 2 à 5).
Figure 1. Déformation des doigts en col de cygne chez une patiente SED hypermobile.
Figure 2. Hyperlaxité des doigts chez une patiente SED hypermobile.
Figure 3. Hyperlaxité du pouce chez une patiente SED hypermobile.
Figure 4. Recuvartum de genou chez une patiente SED hypermobile.
Figure 5. Recuvartum de coude chez un enfant SED hypermobile.
Les enfants peuvent présenter des troubles de la marche et une diminution de leurs capacités physiques et sportives, nécessitant davantage de repos. Les douleurs peuvent être articulaires, musculaires, neuropathiques, mais aussi abdominales, pelviennes ou à type de céphalées. Elles peuvent conduire à un tableau d’hyperalgésie généralisée. Les SED ont des conséquences psychosociales pouvant conduire à des syndromes anxio-dépressifs, à des attitudes d’évitement de certaines situations à risque de récidive d’entorses ou de luxations jusqu’à la kinésiophobie. D’autres symptômes ont été décrits dans le SED hypermobile : asthénie chronique, troubles de la proprioception, maladresse et troubles de la coordination, syndrome de déficit postural, signes fonctionnels musculaires, symptômes digestifs (troubles fonctionnels intestinaux, hernies et prolapsus), troubles vésico-sphinctériens, anomalies rachidiennes, résistance aux anesthésiques locaux, etc.
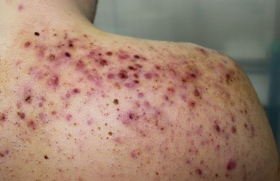
• Le SED classique est dû à des mutations dans les gènes codant pour le collagène 5. Il se manifeste par une atteinte cutanée typique avec cicatrices papyracées atrophiques, hématomes, déchirures cutanées (figure 6). Le SED avec déficit en ténascine X se distingue de la forme classique par sa transmission autosomique récessive, l’absence de cicatrices atrophiques, le risque de fragilité tissulaire et vasculaire, et l’atteinte musculaire.
Figure 6. Séquelle d’hématome chez un patient SED classique.
• Le SED vasculaire est dû à des mutations dans le gène du collagène 3. Les patients ont souvent un morphotype évocateur, une peau fine laissant apparaître le réseau veineux sous-cutané. Ils sont exposés à un risque de complications artérielles de type dissection, anévrisme ou fistule artério-veineuse, à des perforations coliques et à une fragilité de la paroi utérine (en particulier lors de l’accouchement), pouvant engager le pronostic vital.
• Le SED cyphoscoliotique, de transmission autosomique récessive, est dû à un déficit en lysylhydroxylase 1. Il est responsable d’une hypotonie néonatale, d’une cyphoscoliose congénitale sévère et d’un risque de fragilité oculaire pouvant aller du décollement de rétine à la rupture du globe. Par ailleurs, les patients peuvent présenter des cicatrices atrophiques et des complications artérielles.
• Le SED arthrochalasique est dû à des mutations dans les gènes du collagène 1, responsables d’une perte du site de clivage de la procollagène N-propeptide protéinase. Les patients présentent une luxation congénitale des hanches, une hyperlaxité articulaire compliquée d’entorses et de luxations, une fragilité cutanée, un morphotype évocateur et une petite taille.
• Le SED dermatosparaxis, de transmission autosomique récessive est dû à un déficit de l’enzyme chargée du clivage du procollagène de type I (codée par le gène ADAMTS2).
Il se manifeste par une atteinte cutanée majeure, des hernies abdominales, une petite taille et un morphotype évocateur.
• Le SED FKBP22, de transmission autosomique récessive. Il est dû à une mutation du gène FKBP14 codant pour une enzyme qui catalyse le repliement du collagène 3 dans le réticulum endoplasmique. Il est responsable d’une hypotonie musculaire néonatale, d’une myopathie, d’une scoliose évolutive et d’une surdité. Il expose à un risque vasculaire.
• Quatre autres types de SED sont liés à un déficit en enzymes impliquées dans la biosynthèse des protéoglycanes : déficit en galactosyltransférase de type II (codée par le gène B3GALT6), responsable d’une cyphoscoliose sévère, d’une déficience intellectuelle et d’une dysplasie spondylo épimétaphysaire avec fragilité osseuse, déficit en galactosyltransférase de type I (codée par le gène B4GALT7) responsable d’anomalies osseuses et en particulier d’une synostose radio-ulnaire, d’un déficit en dermatane 4-Osulfotransférase 1 (codée par le gène D4ST1), responsable de troubles ophtalmologiques, d’une arthro grypose congénitale et d’un retard psychomoteur, déficit en dermatane sulfate épimérase-1 (codée par le gène DSE1) responsable d’une faiblesse musculaire généralisée, d’une arachnodactylie et de pouces adductus. Ces quatre types de SED sont hérités selon le mode auto somique récessif.
• Le Brittle cornea syndrome est une forme de SED de transmission autosomique récessive due à des mutations dans les gènes ZNF469et PRDM5. Il se manifeste par des signes ophtalmologiques sévères, une surdité et des troubles musculo-squelettiques.
• Le SED spondylocheirodysplasique, de transmission autosomique récessive, est dû à des mutations dans le gène SLC39A13. Les patients présentent un retard de croissance staturale postnatale, une atteinte cutanée, des déformations osseuses et des anomalies dentaires.
• Parmi les autres SED décrits : SED classique avec fragilité vasculaire par mutation du gène COL1A1, SED avec atteinte valvulaire cardiaque par mutation du gène COL1A2 conduisant à l’absence totale de la chaîne alpha, SED avec myopathie par mutation du gène COL12A1 ; SED avec parodontopathie associé à un risque accru d’infections, par mutations dans les gènes C1R et C1S.
Diagnostic
Le diagnostic de SED repose donc sur des critères diagnostiques précis pour chaque sous-type. Plusieurs diagnostics différentiels doivent être éliminés avant de conclure à un SED : syndrome d’hypermobilité articulaire (diagnostiqué grâce aux critères de Beighton et parfois difficile à distinguer du SED hypermobile) ; fibromyalgie ; pathologies rhumatologiques inflammatoires ou auto-immunes ; hétérotopies nodulaires périventriculaires ; myopathies avec hyperlaxité articulaire ; ostéogenèse imparfaite ; syndrome de Marfan ; syndrome de Loeys-Dietz ; syndrome de Silverman, etc. Devant une hyperlaxité associée à une déficience intellectuelle, il convient d’évoquer également d’autres pathologies génétiques syndromiques.
Prise en charge
Les douleurs dans les SED sont complexes : nociceptives, neuropathiques et psychogènes, souvent généralisées et mal soulagées par les antalgiques usuels conduisant les patients à développer des stratégies d’adaptation. Le recours à des antalgiques puissants est fréquent. D’autres méthodes sont utilisées : hypnose, sophrologie, relaxation, acupuncture, mésothérapie, application de patchs antalgiques, de chaleur ou de froid, neurostimulation électrique transcutanée, vêtements compressifs sur mesure.
La prise en charge repose aussi sur la médecine physique et de réadaptation. La kinésithérapie consiste en un travail postural, des exercices faisant travailler la proprioception, la musculation en isométrique, la réassurance et l’éducation. Les manipulations doivent être évitées. La chirurgie orthopédique comporte des risques et doit être discutée au cas par cas (risque de récidive, de saignement, de troubles de la cicatrisation, d’infection, d’adhérences et de majoration des douleurs). Une prise en charge multidisciplinaire est souvent nécessaire chez ces patients. Sur le plan cutané, il convient de protéger les angles du mobilier par des coins mousse et de prescrire des protections de type genouillères, coudières, protègetibias, etc. aux enfants en particulier lors des activités sportives lorsqu’ils présentent une fragilité cutanée. Les sutures doivent comporter davantage de points, avec du fil non résorbable qui sera laissé plus longtemps.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :